library("ggplot2")
library("gridExtra")
library("ggpubr")
library("RColorBrewer")Detect selection in populations
RELATE
AFRICAN POPULATIONS
LWK <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/LWK/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(LWK)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781642 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.1336880 | -0.1734100 | -0.2221450 | -0.3321430 | -0.3804730 | -2.01040e-06 | -0.000017053 | 0 | -0.450077 | -0.0978628 |
| 2 | 2781865 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -2.4857200 | -3.0492300 | -1.8755900 | -2.8888600 | -2.4228300 | -1.39308e+00 | -1.576160000 | 0 | -2.019680 | -1.8051100 |
| 3 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0954429 | -0.0623195 | -0.0632732 | -0.1635510 | -0.4259450 | -7.05956e-02 | -0.034994400 | 0 | -0.228440 | -0.5814090 |
| 4 | 2782572 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.1458270 | -0.2778760 | -0.3514220 | -0.0907462 | -0.0452048 | -1.40217e-01 | -0.176239000 | 0 | -0.167312 | -0.0570220 |
| 5 | 2783658 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 1.0000000 | 1.0000000 | 1.0000000 | 1.0000000 | 1.0000000 | -4.91135e-01 | -0.676691000 | 0 | -1.409400 | -0.4134500 |
| 6 | 2784657 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.1243950 | -0.1766240 | -0.4322030 | -0.4919260 | -0.6999360 | -7.43392e-01 | -0.332010000 | 0 | -0.212519 | -1.0023100 |
LWK_plot <- plot(LWK$pos, -(LWK$when_mutation_has_freq2), pch = 19, col = "#8DA0CB", xlab = "Genomic Position", ylab = "log10(p-value)", main = "LWK",cex = 1.5, yaxp = c(0, 10, 5),cex.lab = 1.2)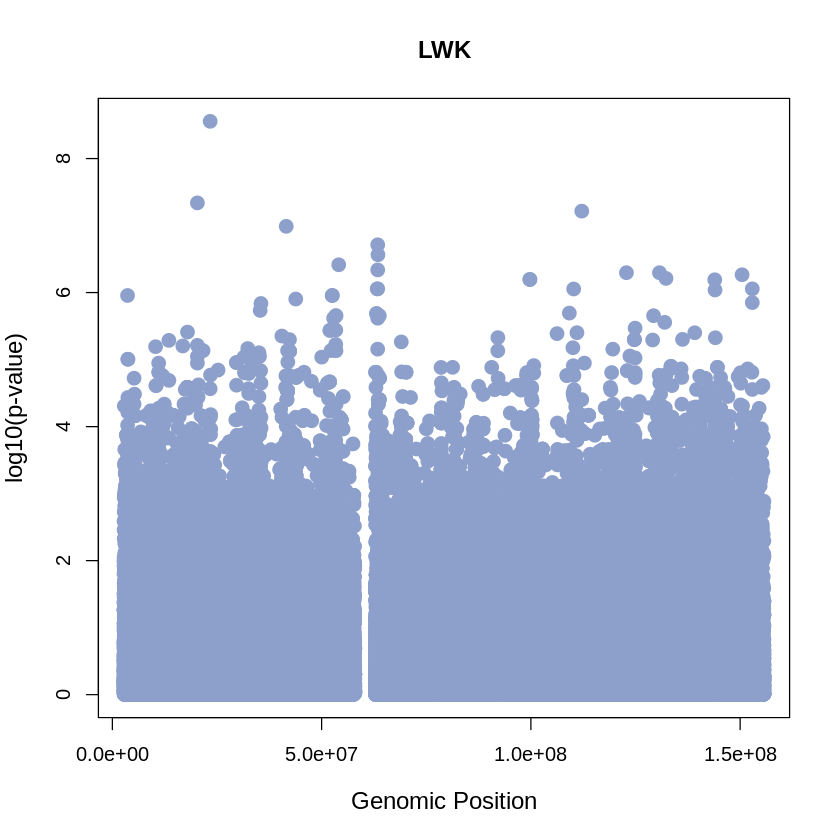
pdf("LWK_plot.pdf", width = 20, height = 5)
LWK_plot <- plot(LWK$pos, -(LWK$when_mutation_has_freq2),
pch = 19, col = "#8DA0CB",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "LWK",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
ESN <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/ESN/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(ESN)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781514 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.241670 | -0.163302 | -0.211304 | -0.385895 | -0.654856 | -0.342752 | -0.492519 | 0 | -0.418871 | -0.757029 |
| 2 | 2781584 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.327150 | -0.196983 | -0.174943 | -0.153555 | -0.248025 | -0.261389 | -0.445471 | 0 | -0.293212 | -0.658046 |
| 3 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.486129 | -0.319048 | -0.511543 | -0.744948 | -0.701433 | -0.315941 | -0.226448 | 0 | -0.849279 | -0.203300 |
| 4 | 2781642 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.355773 | -0.505792 | -0.598037 | -0.100054 | -0.162419 | -0.290461 | -0.382397 | 0 | -0.320757 | -0.190161 |
| 5 | 2781865 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.584338 | -0.299828 | -0.218171 | -0.373706 | -0.187458 | -0.249755 | -0.502119 | 0 | -0.312090 | -0.580090 |
| 6 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.600341 | -0.785731 | -0.495711 | -0.520437 | -0.477516 | -1.343730 | -1.212490 | 0 | -0.488977 | -0.879552 |
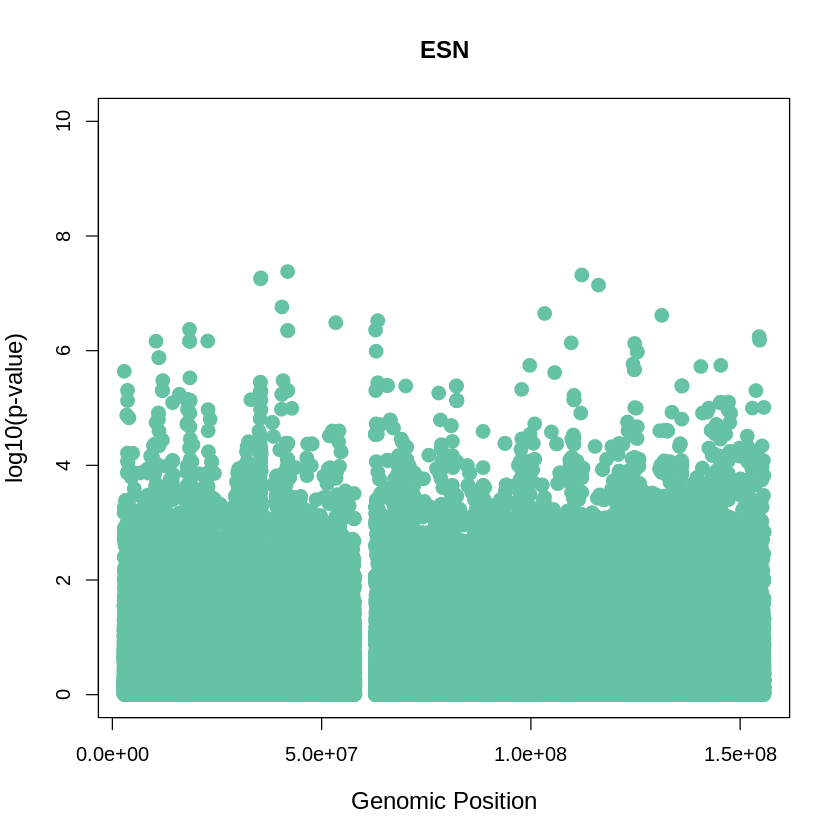
ESN_plot <- plot(ESN$pos, -(ESN$when_mutation_has_freq2), pch=19, col = "#66C2A5", xlab="Genomic Position", ylab="log10(p-value)", main ="ESN", cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("ESN_plot.pdf", width = 20, height = 5)
ESN_plot <- plot(ESN$pos, -(ESN$when_mutation_has_freq2),
pch = 19, col = "#66C2A5",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "ESN",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
GWD <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/GWD/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(GWD)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781514 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.407876 | -0.463329 | -0.743589 | -0.597593 | -0.507644 | -0.253240 | -0.349971000 | 0 | -0.614516 | -0.656961 |
| 2 | 2781584 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.179203 | -0.154162 | -0.180094 | -0.205703 | -0.124971 | -0.231529 | -0.204117000 | 0 | -0.240526 | -0.268348 |
| 3 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.111829 | -0.212617 | -0.202914 | -0.378425 | -0.322488 | -0.277704 | -0.282409000 | 0 | -0.190963 | -0.782312 |
| 4 | 2781642 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 1.000000 | -0.539265 | -0.347216 | -0.565445 | -0.308591 | -0.153304 | -0.000101637 | 0 | -0.686005 | -0.379514 |
| 5 | 2781865 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.519564 | -0.462069 | -0.316058 | -0.352527 | -0.137388 | -0.134281 | -0.264966000 | 0 | -0.590395 | -0.857276 |
| 6 | 2782386 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.402728 | -0.221762 | -0.361282 | -0.213686 | -0.360681 | -0.502329 | -0.307497000 | 0 | -0.555433 | -0.201529 |
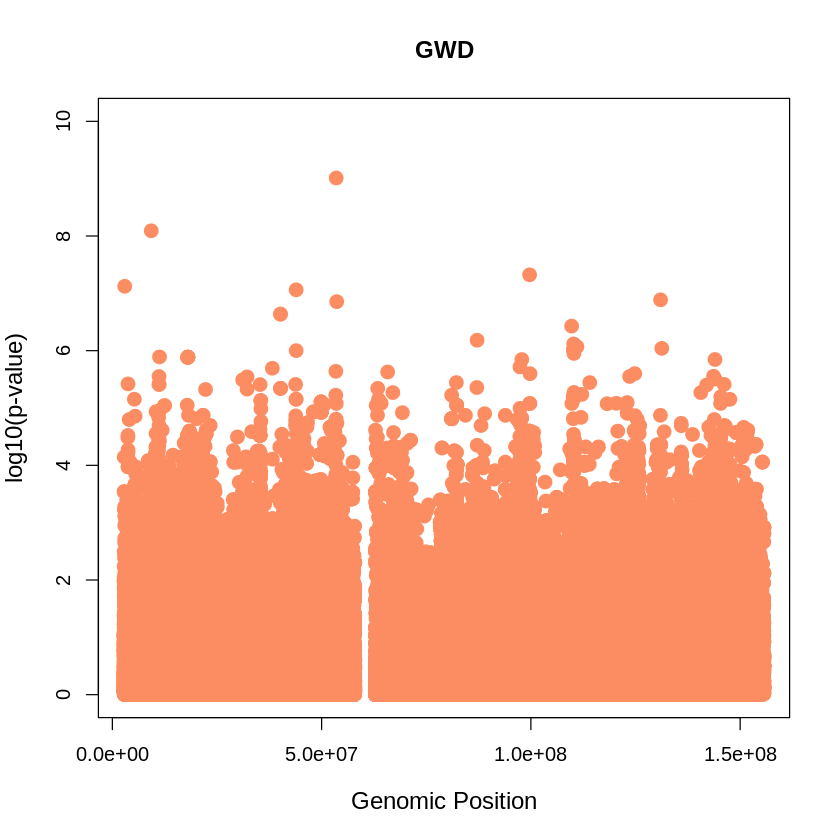
GWD_plot <- plot(GWD$pos, -(GWD$when_mutation_has_freq2), pch = 19, col = "#FC8D62", xlab = "Genomic Position", ylab = "log10(p-value)", main = "GWD", cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("GWD_plot.pdf", width = 20, height = 5)
GWD_plot <- plot(GWD$pos, -(GWD$when_mutation_has_freq2),
pch = 19, col = "#FC8D62",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "GWD",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
MSL <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/MSL/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(MSL)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781514 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.00819344 | -0.0230286 | -0.0125319 | -0.03071400 | -0.0335299 | -0.0137678 | -0.0461147 | 0 | -0.0346441 | -0.1966720 |
| 2 | 2781584 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.57859900 | -0.6665050 | -0.4813950 | -0.19035200 | -0.0996204 | -0.3150730 | -0.0203248 | 0 | -0.6003820 | -0.3105280 |
| 3 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.48882200 | -0.6708420 | -0.4792100 | -0.46056600 | -0.7251980 | -0.1401750 | -0.2732000 | 0 | -0.7939040 | -1.4443700 |
| 4 | 2781865 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.72332800 | -0.4880510 | -0.3577220 | -0.49751700 | -0.1608950 | -0.2124540 | -0.3456440 | 0 | -0.7253440 | -0.2675890 |
| 5 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.40212600 | -0.1816380 | -0.2759440 | -0.20043600 | -0.1026650 | -0.1161240 | -0.1489970 | 0 | -0.4218150 | -0.4057530 |
| 6 | 2783658 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.06384770 | -0.0422283 | -0.0282915 | -0.00793583 | -0.0163262 | -0.0427755 | 0.0000000 | 0 | -0.0417048 | -0.0653668 |
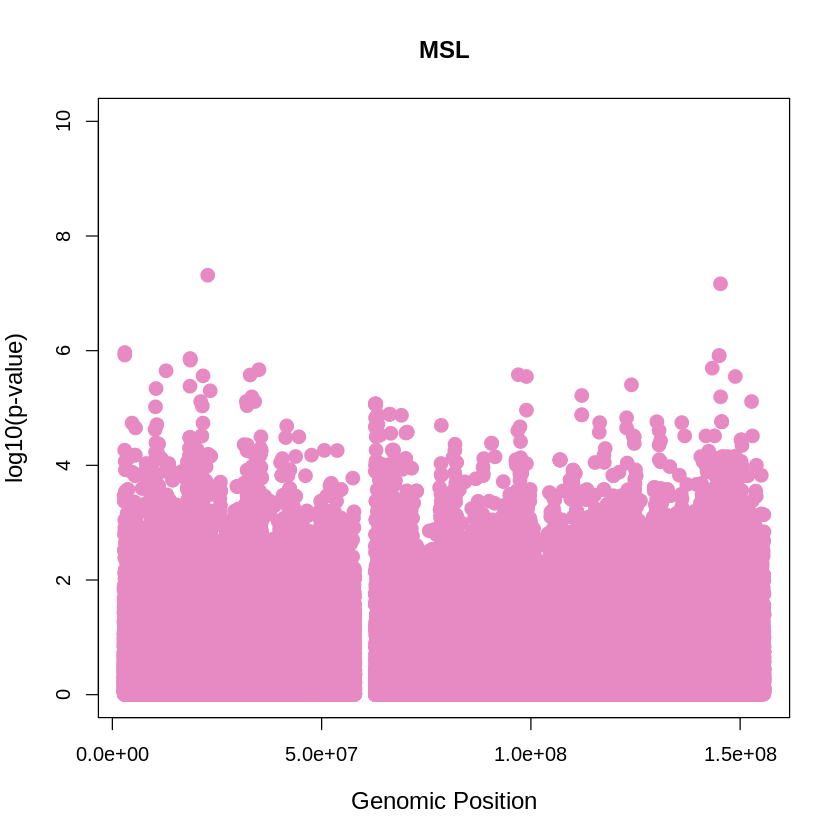
MSL_plot <- plot(MSL$pos, -(MSL$when_mutation_has_freq2), pch = 19, col = "#E78AC3", xlab = "Genomic Position", ylab = "log10(p-value)", main = "MSL", cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("MSL_plot.pdf", width = 20, height = 5)
MSL_plot <- plot(MSL$pos, -(MSL$when_mutation_has_freq2),
pch = 19, col = "#E78AC3",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "MSL",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
YRI <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/YRI/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(YRI)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781584 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.202970 | -0.649106 | -1.2164900 | -0.6031470 | -0.860137 | -1.338100 | -1.0011900 | 0 | -1.181280 | -0.5485560 |
| 2 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.174386 | -0.318978 | -0.0721344 | -0.0799852 | -0.184531 | -0.190856 | -0.3960370 | 0 | -0.102744 | -0.5156580 |
| 3 | 2781642 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 1.000000 | 1.000000 | 1.0000000 | 1.0000000 | -0.621146 | -0.869970 | -1.2066100 | 0 | -1.387990 | -0.6117740 |
| 4 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.668357 | -0.674931 | -0.6905980 | -0.5737620 | -0.274520 | -0.216861 | -0.3180610 | 0 | -0.517044 | -1.2772800 |
| 5 | 2782597 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.262238 | -0.398503 | -0.5650450 | -0.6882240 | -0.131008 | -0.191674 | -0.0770765 | 0 | -0.399880 | -0.2131010 |
| 6 | 2783658 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.514807 | -0.961053 | -0.9117400 | -0.7238790 | -1.248990 | -1.566840 | -1.3695800 | 0 | -1.302540 | -0.0112159 |
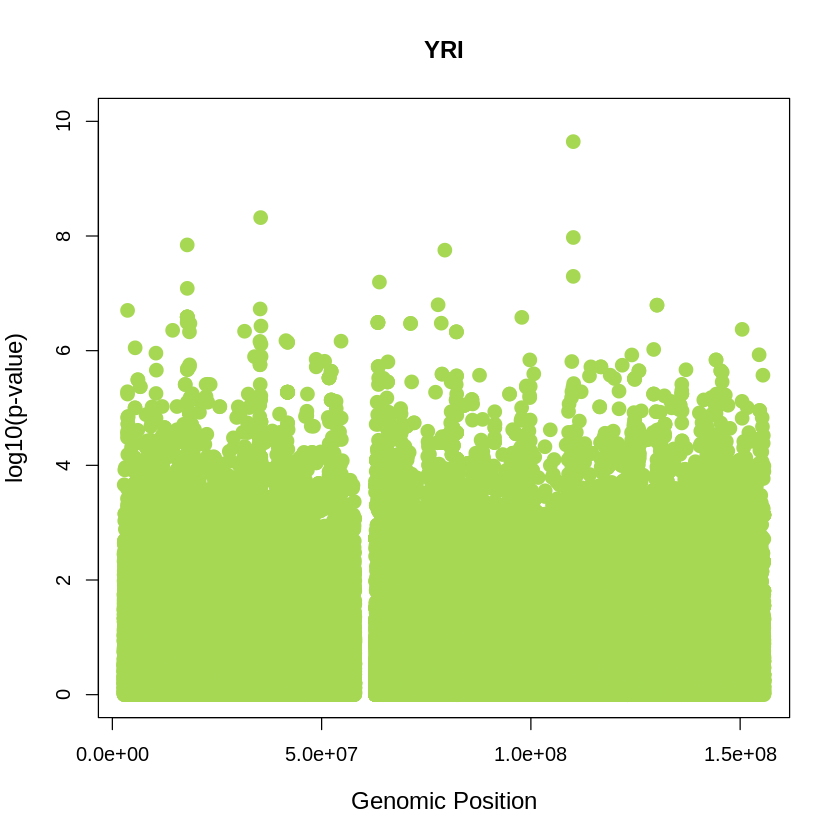
YRI_plot <- plot(YRI$pos, -(YRI$when_mutation_has_freq2), pch = 19, col = "#A6D854", xlab = "Genomic Position", ylab = "log10(p-value)", main = "YRI" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("YRI_plot.pdf", width = 20, height = 5)
YRI_plot <- plot(YRI$pos, -(YRI$when_mutation_has_freq2),
pch = 19, col = "#A6D854",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "YRI",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
# Combine data frames
LWK$Population <- "LWK"
ESN$Population <- "ESN"
GWD$Population <- "GWD"
MSL$Population <- "MSL"
YRI$Population <- "YRI"
combined_data <- rbind(LWK, ESN, GWD, MSL, YRI)
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = -(when_mutation_has_freq2), color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "log10(p-value)", title = "Africans") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_brewer(palette = "Set2") +
theme(
text = element_text(size = 15),
plot.title = element_text(hjust = 0.5) # Center the title
)
# Print the plot
print(plot_combined)
# Save the plot
ggsave("africans_threshold_REL_SQUARE.png", plot_combined, width = 7, height = 5)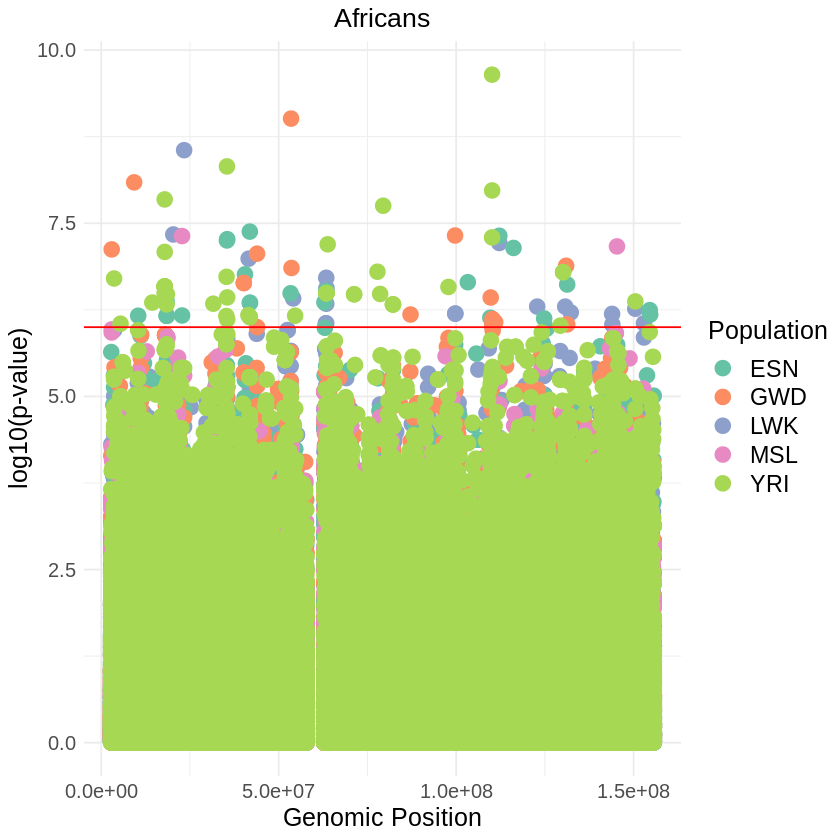
## SELECT JUST POSITION LOWER THAN THRESHOLD (log10 p-value < -6)
# Define file paths for the five .sele files
file_paths <- c(
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/LWK/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/ESN/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/GWD/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/MSL/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/YRI/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele"
)
# Read the files
LWK <- read.table(file_paths[1], header = TRUE)
ESN <- read.table(file_paths[2], header = TRUE)
GWD <- read.table(file_paths[3], header = TRUE)
MSL <- read.table(file_paths[4], header = TRUE)
YRI <- read.table(file_paths[5], header = TRUE)
# Function to subset data based on log10 p-value
subset_data <- function(data, population) {
subset(data, when_mutation_has_freq2 < -6) %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = LWK, population = "LWK"),
list(data = ESN, population = "ESN"),
list(data = GWD, population = "GWD"),
list(data = MSL, population = "MSL"),
list(data = YRI, population = "YRI")
)
# Subset and bind data from all files
africans_low_pvalue <- do.call(rbind, lapply(file_pop, function(fp) {
subset_data(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(africans_low_pvalue, "africans_low_pvalue.csv", row.names = FALSE)
filtered <- read.csv("/home/ari/ari-intern/people/ari/ariadna-intern/notebooks/africans_low_pvalue.csv")
head(filtered)
cat("Number of rows and columns:", paste(dim(filtered), collapse = "x"))plot_threshold <- ggplot(filtered, aes(x = pos, y = -(when_mutation_has_freq2), color = population)) +
geom_point(size = 4) +
labs(x = "Genomic position", y = "log10 p-value", title = "AFRICAN") +
theme_minimal() +
scale_color_brewer(palette = "Set2") + theme(text = element_text(size = 15),
plot.title = element_text(hjust = 0.5))
print(plot_threshold)
ggsave("africans_low_pvalue_r.png", plot_threshold, width = 20, height = 8)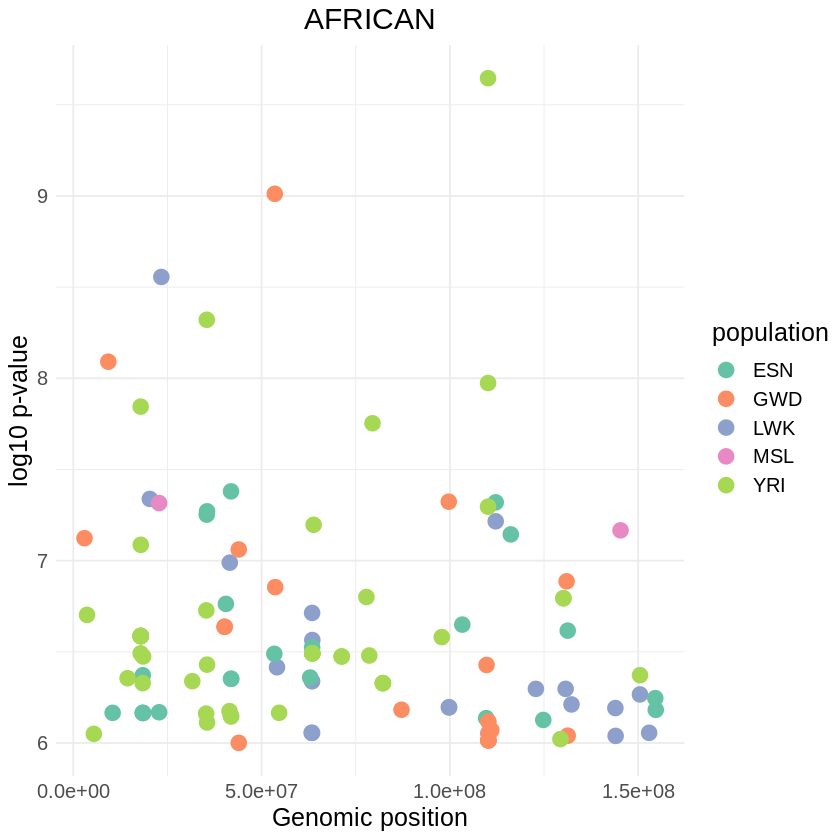
EUROPEAN POPULATIONS
GBR <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/GBR/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(GBR)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781604 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.04246550 | -0.0410563 | -0.0277191 | 0.00000000 | 0.0000000 | 0.0000000 | 0.000000 | 0 | -0.0652181 | -0.140835 |
| 2 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.07381820 | -0.0622531 | -0.1658510 | -0.20821400 | -0.1090100 | -0.2489250 | 0.000000 | 0 | -0.2119610 | -0.159586 |
| 3 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.63264000 | -1.4569200 | -1.0390600 | -1.39977000 | -1.1803100 | -0.5330060 | -0.624974 | 0 | -2.2223900 | -1.710600 |
| 4 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.63264000 | -1.4569200 | -1.0390600 | -1.39977000 | -1.1803100 | -0.5330060 | -0.624974 | 0 | -2.2223900 | -1.710600 |
| 5 | 2782116 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.06384060 | -0.0555249 | -0.0357912 | 0.00000000 | 0.0000000 | 0.0000000 | 0.000000 | 0 | -0.1008840 | -0.372146 |
| 6 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.00997385 | -0.0102032 | -0.0278379 | -0.00742967 | -0.0071796 | -0.0841723 | 0.000000 | 0 | -0.0321039 | -0.141249 |
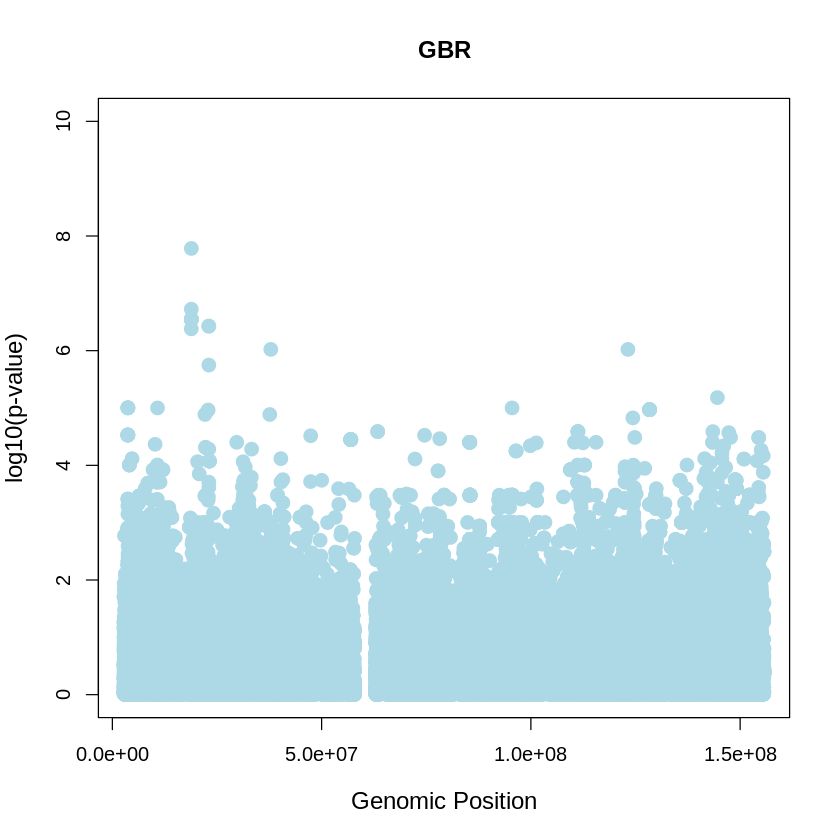
GBR_plot <- plot(GBR$pos, -(GBR$when_mutation_has_freq2), pch = 19, col = "lightblue", xlab = "Genomic Position", ylab = "log10(p-value)", main = "GBR" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("GBR_plot.pdf", width = 20, height = 5)
YRI_plot <- plot(GBR$pos, -(GBR$when_mutation_has_freq2),
pch = 19, col = "lightblue",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "GBR",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
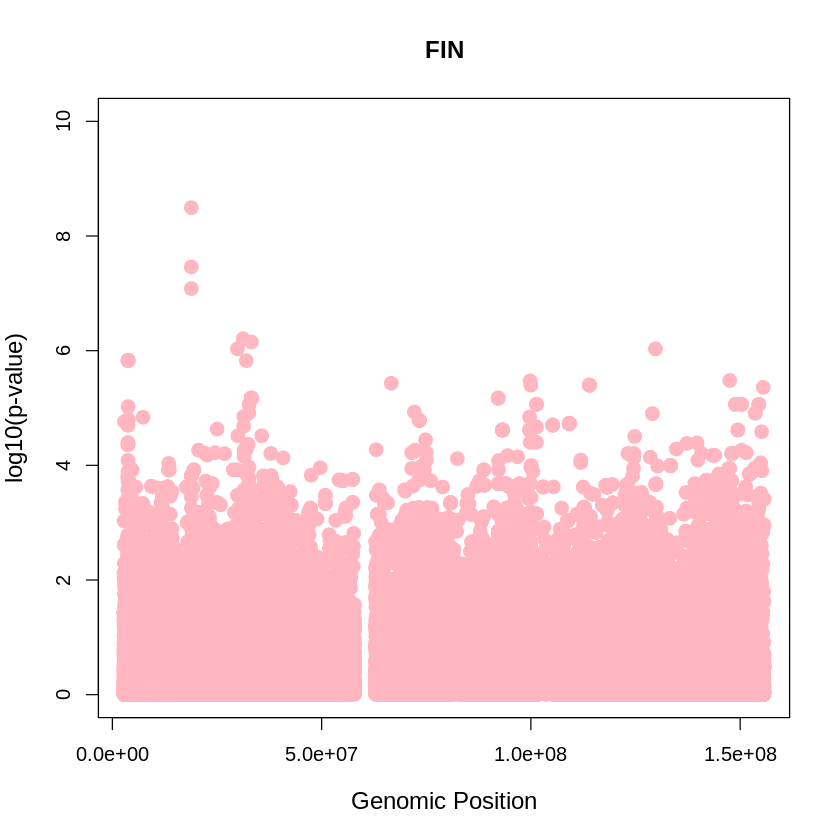
FIN <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/FIN/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(FIN)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781604 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.00440755 | -0.00534391 | -0.0121758 | -0.0395929 | -0.12861100 | -0.07316910 | -4.72098e-05 | 0 | -0.00567415 | -0.0443709 |
| 2 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.40902500 | -0.62795400 | -0.3375140 | -0.4141800 | -0.58893800 | -0.68931200 | -1.13840e+00 | 0 | -0.57942200 | -0.4660740 |
| 3 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.65529300 | -0.20453900 | -0.2242660 | -0.0580616 | -0.00696292 | -0.00380284 | -3.23889e-03 | 0 | -0.45251000 | -1.3097700 |
| 4 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.65529300 | -0.20453900 | -0.2242660 | -0.0580616 | -0.00696292 | -0.00380284 | -3.23889e-03 | 0 | -0.45251000 | -1.3097700 |
| 5 | 2782116 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.00440755 | -0.00534391 | -0.0121758 | -0.0395929 | -0.12861100 | -0.07316910 | -4.72098e-05 | 0 | -0.00567415 | -0.0443709 |
| 6 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.04182390 | -0.09758080 | -0.0624887 | -0.1448340 | -0.37536700 | -0.37688400 | -4.58744e-01 | 0 | -0.03356820 | -0.0363945 |
FIN_plot <- plot(FIN$pos, -(FIN$when_mutation_has_freq2), pch = 19, col = "lightpink", xlab = "Genomic Position", ylab = "log10(p-value)", main = "FIN" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("FIN_plot.pdf", width = 20, height = 5)
YRI_plot <- plot(FIN$pos, -(FIN$when_mutation_has_freq2),
pch = 19, col = "lightpink",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "FIN",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
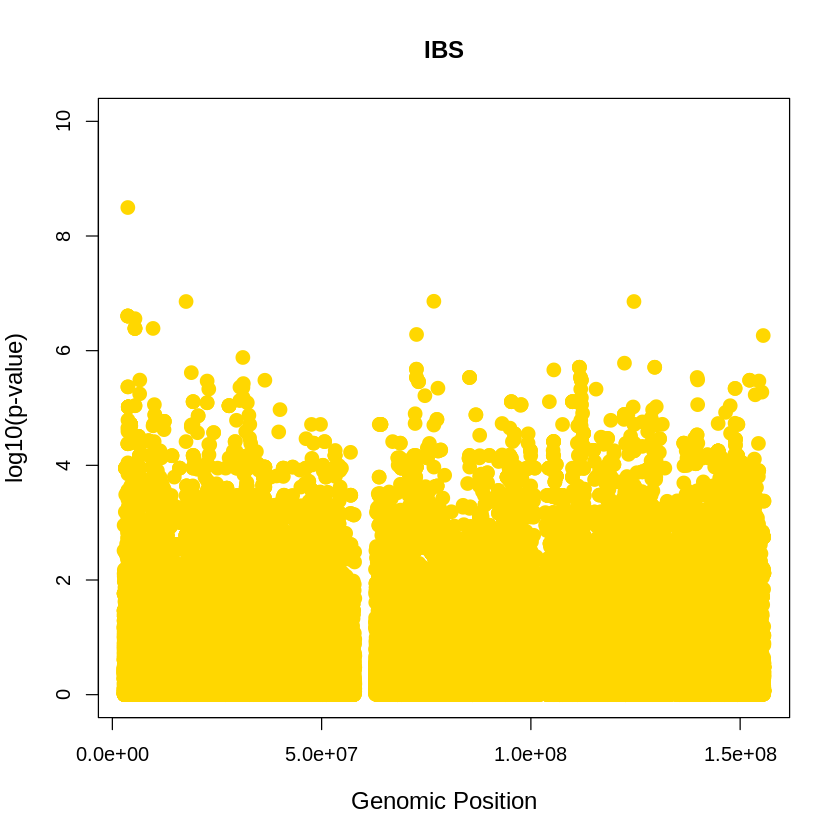
IBS <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/IBS/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(IBS)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781514 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.5030530 | -0.62381400 | -0.6142110 | -0.5986810 | -0.2936090 | -0.5673030 | -0.5812090 | 0 | -0.5659250 | -0.430028 |
| 2 | 2781604 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.1494370 | -0.13499200 | -0.0992126 | -0.0863440 | -0.0437142 | -0.0229475 | -0.0256627 | 0 | -0.0950446 | -0.487534 |
| 3 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.6871990 | -0.41871100 | -0.4638830 | -0.4103800 | -0.7481900 | -0.2844150 | -0.4797510 | 0 | -0.9172860 | -0.408764 |
| 4 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0408422 | -0.00879619 | -0.0348159 | -0.0113192 | -0.0213689 | -0.0237058 | -0.1975070 | 0 | -0.0570564 | -1.165120 |
| 5 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0408422 | -0.00879619 | -0.0348159 | -0.0113192 | -0.0213689 | -0.0237058 | -0.1975070 | 0 | -0.0570564 | -1.165120 |
| 6 | 2782048 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 1.0000000 | 1.00000000 | 1.0000000 | 1.0000000 | 1.0000000 | -0.9076720 | -1.1323700 | 0 | -1.5064100 | -0.838322 |
IBS_plot <- plot(IBS$pos, -(IBS$when_mutation_has_freq2), pch = 19, col = "gold", xlab = "Genomic Position", ylab = "log10(p-value)", main = "IBS" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("IBS_plot.pdf", width = 20, height = 5)
IBS_plot <- plot(IBS$pos, -(IBS$when_mutation_has_freq2),
pch = 19, col = "gold",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "IBS",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
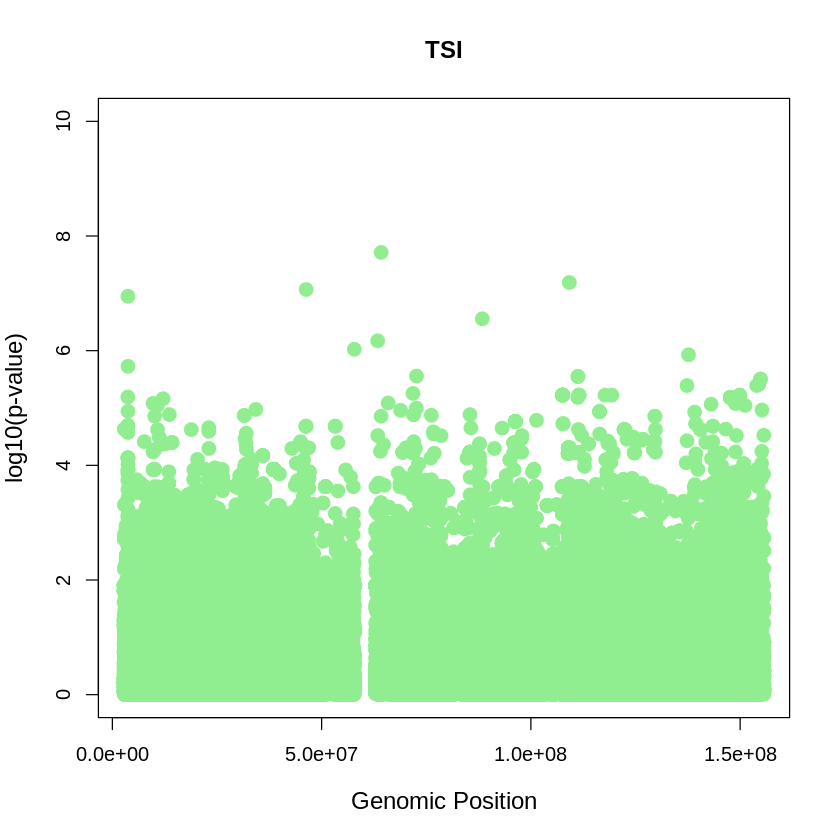
TSI <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/TSI/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(TSI)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781604 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.2105130 | -0.0994744 | -0.00275779 | -0.0264850 | -0.1290760 | -4.82756e-05 | -2.05973e-05 | 0 | -0.239337 | -0.2886860 |
| 2 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0223887 | -0.0254137 | -0.05456920 | -0.0224805 | -0.0427598 | -4.61010e-05 | -2.00428e-05 | 0 | -0.175876 | -0.1633620 |
| 3 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.7507600 | -1.5958100 | -2.17561000 | -1.6023800 | -0.9149070 | -9.32056e-01 | -1.16969e-01 | 0 | -1.454490 | -1.2938300 |
| 4 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.7507600 | -1.5958100 | -2.17561000 | -1.6023800 | -0.9149070 | -9.32056e-01 | -1.16969e-01 | 0 | -1.454490 | -1.2938300 |
| 5 | 2782116 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.1580850 | -0.0765467 | -0.00201294 | -0.0222592 | -0.1175840 | -3.60871e-05 | -1.89802e-05 | 0 | -0.210797 | -0.0313601 |
| 6 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.3088920 | -0.2460760 | -0.54315200 | -0.2379020 | -0.5093210 | -5.29402e-01 | -1.07703e-01 | 0 | -0.446141 | -0.3001710 |
TSI_plot <- plot(TSI$pos, -(TSI$when_mutation_has_freq2), pch = 19, col = "lightgreen", xlab = "Genomic Position", ylab = "log10(p-value)", main = "TSI" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("TSI_plot.pdf", width = 20, height = 5)
TSI_plot <- plot(TSI$pos, -(TSI$when_mutation_has_freq2),
pch = 19, col = "lightgreen",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "TSI",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
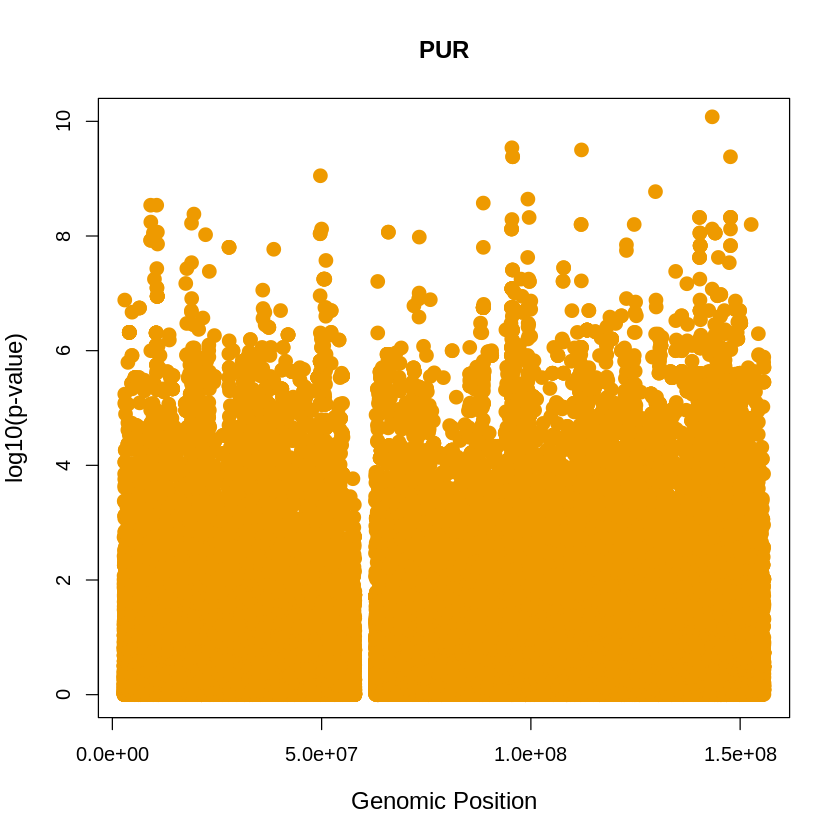
PUR <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/PUR/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
PUR_plot <- plot(PUR$pos, -(PUR$when_mutation_has_freq2), pch = 19, col = "orange2", xlab = "Genomic Position", ylab = "log10(p-value)", main = "PUR" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("PUR_plot.pdf", width = 20, height = 5)
PUR_plot <- plot(PUR$pos, -(PUR$when_mutation_has_freq2),
pch = 19, col = "orange2",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "PUR",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
# Combine data frames
GBR$Population <- "GBR"
FIN$Population <- "FIN"
IBS$Population <- "IBS"
TSI$Population <- "TSI"
PUR$Population <- "PUR"
combined_data <- rbind(GBR, FIN, IBS, TSI, PUR)
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = -(when_mutation_has_freq2), color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "log10(p-value)", title = "Europeans + PUR") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_manual(values = c("GBR" = "lightblue", "FIN" = "lightpink", "IBS" = "gold", "TSI" = "lightgreen", "PUR" = "orange2")) + theme(text = element_text(size = 15))
print(plot_combined)
ggsave("europeans_threshold_REL_SQUARE.png", plot_combined, width = 7, height = 5)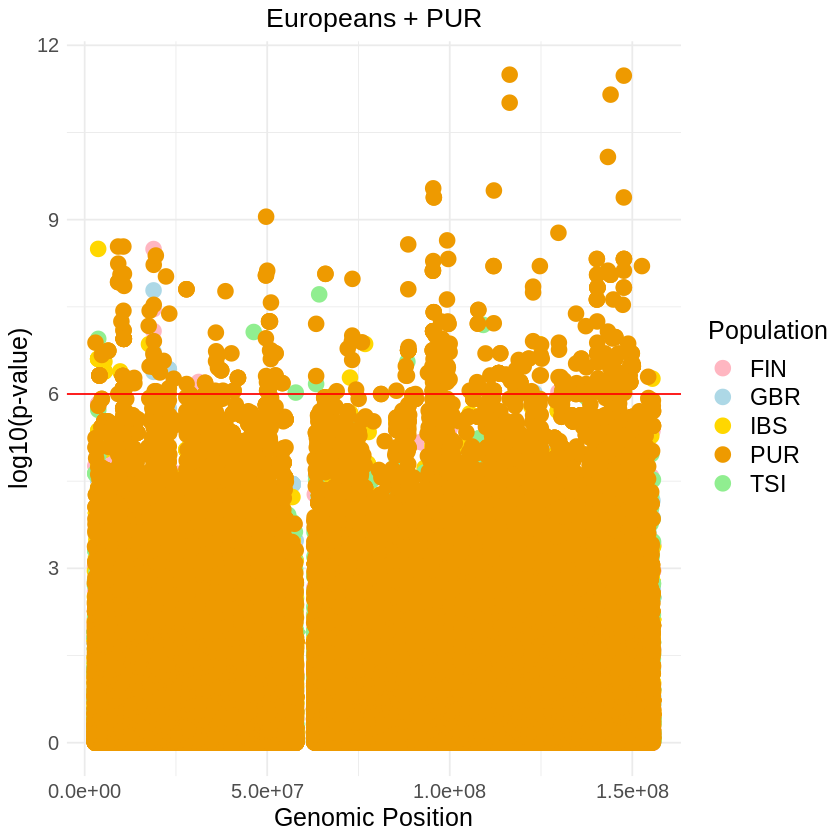
## SELECT JUST POSITION LOWER THAN THRESHOLD (log10 p-value < -6)
# Define file paths for the five .sele files
file_paths_euro <- c(
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/GBR/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/FIN/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/IBS/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/TSI/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/PUR/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele"
)
# Read the files
GBR <- read.table(file_paths_euro[1], header = TRUE)
FIN <- read.table(file_paths_euro[2], header = TRUE)
IBS <- read.table(file_paths_euro[3], header = TRUE)
TSI <- read.table(file_paths_euro[4], header = TRUE)
PUR <- read.table(file_paths_euro[5], header = TRUE)
# Function to subset data based on log10 p-value
subset_data <- function(data, population) {
subset(data, when_mutation_has_freq2 < -6) %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = GBR, population = "GBR"),
list(data = FIN, population = "FIN"),
list(data = IBS, population = "IBS"),
list(data = TSI, population = "TSI"),
list(data = PUR, population = "PUR")
)
# Subset and bind data from all files
europeans_low_pvalue <- do.call(rbind, lapply(file_pop, function(fp) {
subset_data(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(europeans_low_pvalue, "europeans_low_pvalue.csv", row.names = FALSE)
filtered_euro <- read.csv("/home/ari/ari-intern/people/ari/ariadna-intern/notebooks/europeans_low_pvalue.csv")
head(filtered_euro)
cat("Number of rows and columns:", paste(dim(filtered_euro), collapse = "x"))| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | population | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <int> | <int> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | <chr> | |
| 1 | 18861677 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.597739 | -1.048370 | -0.741127 | -0.417716 | -0.544644 | -0.187459 | 0 | -0.890654 | -6.38066 | GBR |
| 2 | 18865240 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.639418 | -0.897673 | -0.582284 | -0.612257 | -0.702097 | -0.241534 | 0 | -0.991791 | -6.72386 | GBR |
| 3 | 18867826 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.137600 | -0.715037 | -0.976634 | -0.572180 | -0.612715 | -0.132271 | 0 | -1.339690 | -6.54745 | GBR |
| 4 | 18867954 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.127090 | -0.618784 | -1.215980 | -0.720713 | -0.718903 | -0.155134 | 0 | -1.423040 | -7.78284 | GBR |
| 5 | 18868013 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.137600 | -0.715037 | -0.976634 | -0.572180 | -0.612715 | -0.132271 | 0 | -1.339690 | -6.54745 | GBR |
| 6 | 18868321 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.137600 | -0.715037 | -0.976634 | -0.572180 | -0.612715 | -0.132271 | 0 | -1.339690 | -6.54745 | GBR |
Number of rows and columns: 456x36plot_threshold <- ggplot(filtered_euro, aes(x = pos, y = -(when_mutation_has_freq2), color = population)) +
geom_point(size = 4) +
labs(x = "Genomic position", y = "log10 p-value", title = "European Populations") +
theme_minimal() + scale_color_manual(values = c("GBR" = "lightblue", "FIN" = "lightpink", "IBS" = "gold", "TSI" = "lightgreen", "PUR" = "orange2")) +
theme(text = element_text(size = 15),
plot.title = element_text(hjust = 0.5))
print(plot_threshold)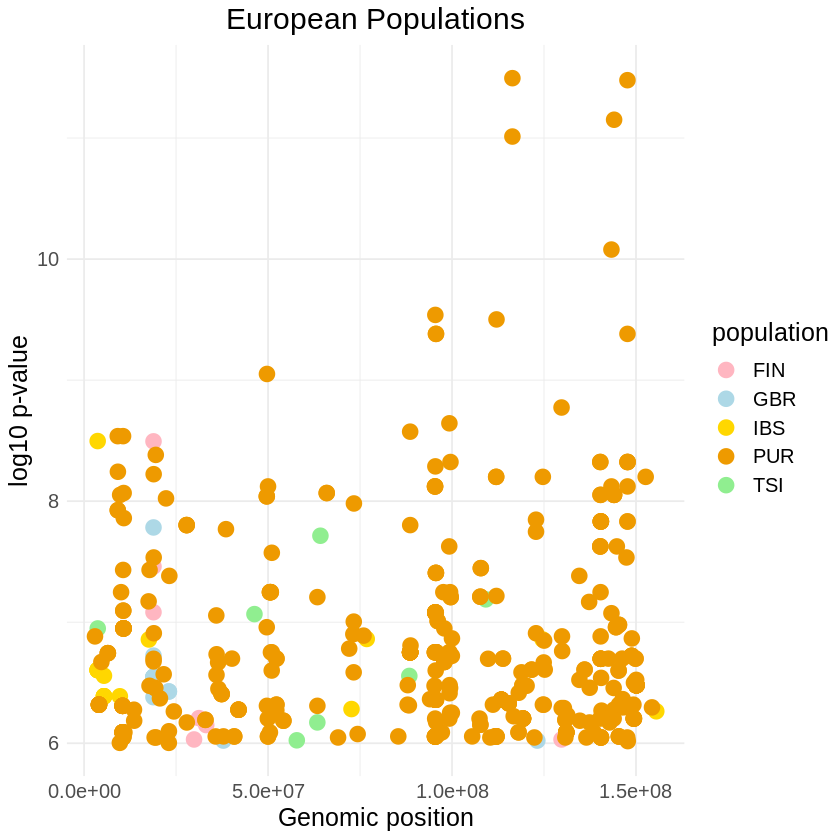
EAST ASIAN POPULATIONS
CDX <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/CDX/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(CDX)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.0086400 | -1.1085600 | -1.1057000 | -1.3585700 | -0.943955 | -1.1108200 | -0.973984 | 0 | -1.2454700 | -0.4867200 |
| 2 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.3131650 | -0.2109630 | -0.2253310 | -0.0785041 | -0.164815 | -0.1097100 | -0.108324 | 0 | -0.3075960 | -0.5534800 |
| 3 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.3131650 | -0.2109630 | -0.2253310 | -0.0785041 | -0.164815 | -0.1097100 | -0.108324 | 0 | -0.3075960 | -0.5534800 |
| 4 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.0339200 | -1.0921500 | -1.3285200 | -1.5450600 | -1.067340 | -1.2125100 | -1.057640 | 0 | -1.2068300 | -1.3679800 |
| 5 | 2782560 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 0.0000000 | 0.0000000 | 0.0000000 | 0.0000000 | 0.000000 | 0.0000000 | 0.000000 | 0 | -0.0766008 | -0.0269834 |
| 6 | 2782572 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0993169 | -0.0502764 | -0.0654512 | -0.0539695 | -0.238131 | -0.0119095 | -0.033889 | 0 | -0.1106720 | -1.6958700 |
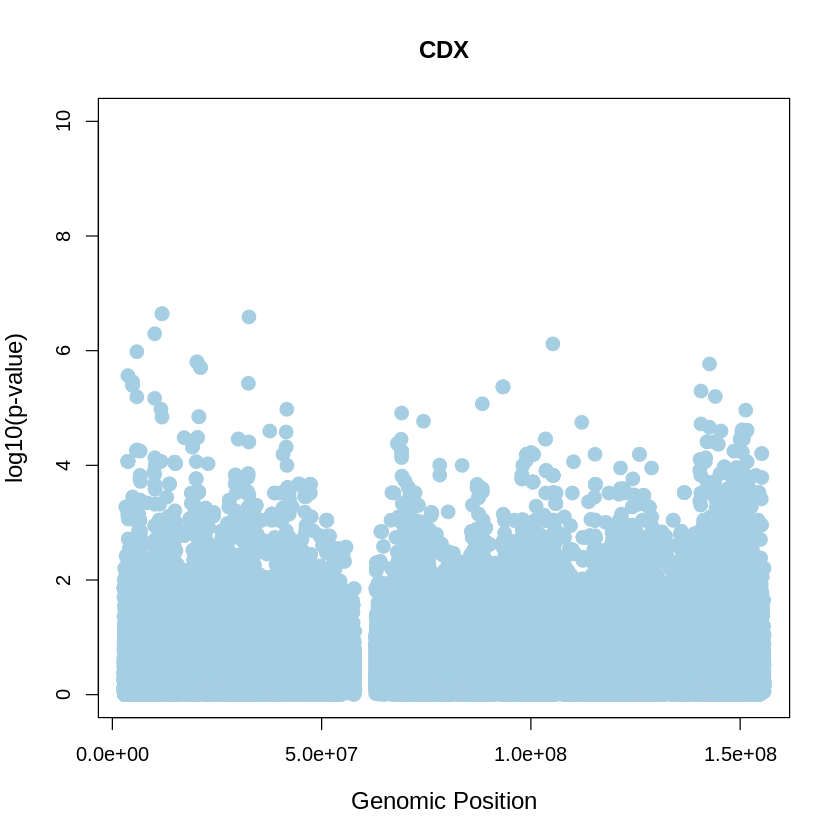
CDX_plot <- plot(CDX$pos, -(CDX$when_mutation_has_freq2), pch = 19, col = "#A6CEE3", xlab = "Genomic Position", ylab = "log10(p-value)", main = "CDX" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("CDX_plot.pdf", width = 20, height = 5)
CDX_plot <- plot(CDX$pos, -(CDX$when_mutation_has_freq2),
pch = 19, col = "#A6CEE3",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "CDX",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
CHB <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/CHB/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(CHB)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.469389 | -0.1350390 | -0.0537125 | -0.0683953 | -0.1218410 | -1.66876e-05 | -4.30725e-07 | 0 | -0.357821 | -0.707989 |
| 2 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.469389 | -0.1350390 | -0.0537125 | -0.0683953 | -0.1218410 | -1.66876e-05 | -4.30725e-07 | 0 | -0.357821 | -0.707989 |
| 3 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.134001 | -0.1969290 | -0.3636970 | -0.1665180 | -0.0778992 | -1.11767e-01 | -1.29266e-06 | 0 | -0.223143 | -0.803470 |
| 4 | 2782572 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.352616 | -0.0748059 | -0.0297824 | -0.1350440 | -0.0487577 | 0.00000e+00 | 0.00000e+00 | 0 | -0.314035 | -2.224800 |
| 5 | 2784758 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.414356 | -0.1018040 | -0.1638560 | -0.1877580 | -0.0405383 | -3.93919e-05 | 0.00000e+00 | 0 | -0.292926 | -0.067107 |
| 6 | 2784905 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.414356 | -0.1018040 | -0.1638560 | -0.1877580 | -0.0405383 | -3.93919e-05 | 0.00000e+00 | 0 | -0.292926 | -0.067107 |
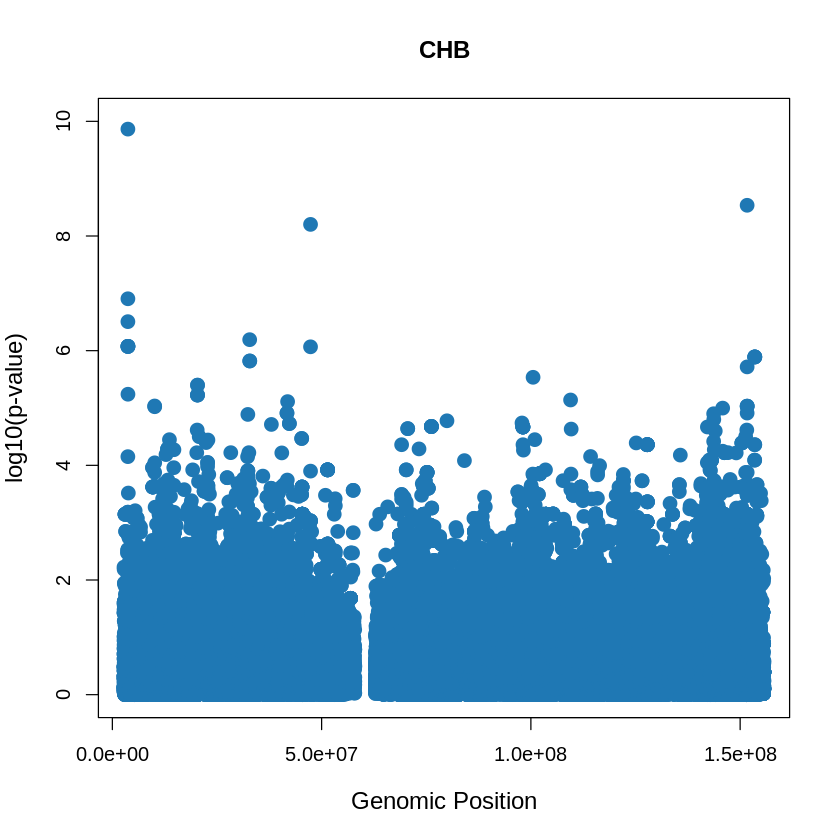
CHB_plot <- plot(CHB$pos, -(CHB$when_mutation_has_freq2), pch = 19, col = "#1F78B4", xlab = "Genomic Position", ylab = "log10(p-value)", main = "CHB" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("CHB_plot.pdf", width = 20, height = 5)
CHB_plot <- plot(CHB$pos, -(CHB$when_mutation_has_freq2),
pch = 19, col = "#1F78B4",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "CHB",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
CHS <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/CHS/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(CHS)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.509651 | -0.5295390 | -0.540640 | -0.2382600 | -0.353003 | -0.495016 | -0.453266 | 0 | -0.6016760 | -0.926999 |
| 2 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.191600 | -0.0350659 | -0.102777 | -0.0583544 | -0.278356 | -0.185307 | -0.293794 | 0 | -0.0503824 | -0.294591 |
| 3 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.191600 | -0.0350659 | -0.102777 | -0.0583544 | -0.278356 | -0.185307 | -0.293794 | 0 | -0.0503824 | -0.294591 |
| 4 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.921153 | -1.5332800 | -0.849335 | -0.7494320 | -0.419384 | -0.937000 | -0.393215 | 0 | -0.9510930 | -1.741520 |
| 5 | 2782572 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.719856 | -0.7079780 | -0.505628 | -0.5949980 | -0.269845 | -0.588185 | -0.232325 | 0 | -0.5436550 | -0.533548 |
| 6 | 2782784 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 1.000000 | 1.0000000 | 1.000000 | 1.0000000 | 1.000000 | 1.000000 | 1.000000 | 0 | 1.0000000 | -0.701893 |
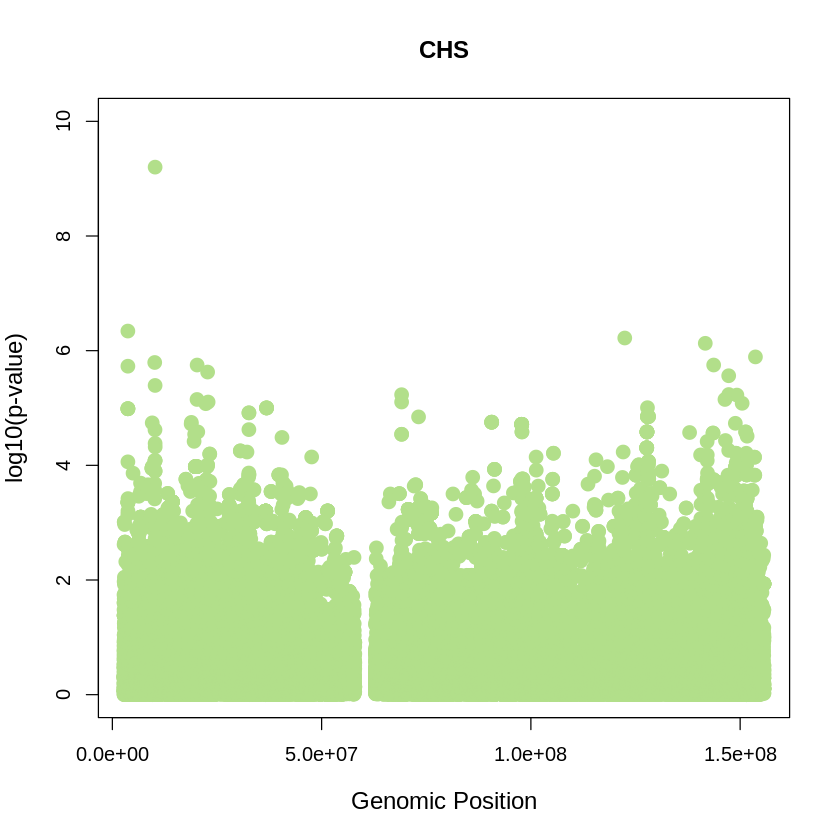
CHS_plot <- plot(CHS$pos, -(CHS$when_mutation_has_freq2), pch = 19, col = "#B2DF8A", xlab = "Genomic Position", ylab = "log10(p-value)", main = "CHS" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("CHS_plot.pdf", width = 20, height = 5)
CHS_plot <- plot(CHS$pos, -(CHS$when_mutation_has_freq2),
pch = 19, col = "#B2DF8A",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "CHS",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
JPT <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/JPT/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(JPT)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781514 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.544881 | -0.543074 | -0.3360220 | -0.5822880 | -0.4354700 | -0.6970980 | -1.1308500 | 0 | -0.548842 | -0.746704 |
| 2 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.354544 | -0.370605 | -0.0512392 | -0.0805736 | -0.0537353 | -0.1730980 | -0.4372470 | 0 | -0.455204 | -0.112781 |
| 3 | 2782572 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.319862 | -0.318994 | -0.4698040 | -0.2719470 | -0.0442616 | -0.0408214 | -0.0908543 | 0 | -0.396086 | -1.069260 |
| 4 | 2784758 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.629078 | -0.263415 | -0.7430090 | -0.3709640 | -0.8436410 | -0.9887650 | -0.7396860 | 0 | -0.796063 | -0.483658 |
| 5 | 2784905 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.629078 | -0.263415 | -0.7430090 | -0.3709640 | -0.8436410 | -0.9887650 | -0.7396860 | 0 | -0.796063 | -0.483658 |
| 6 | 2785313 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.629078 | -0.263415 | -0.7430090 | -0.3709640 | -0.8436410 | -0.9887650 | -0.7396860 | 0 | -0.796063 | -0.483658 |
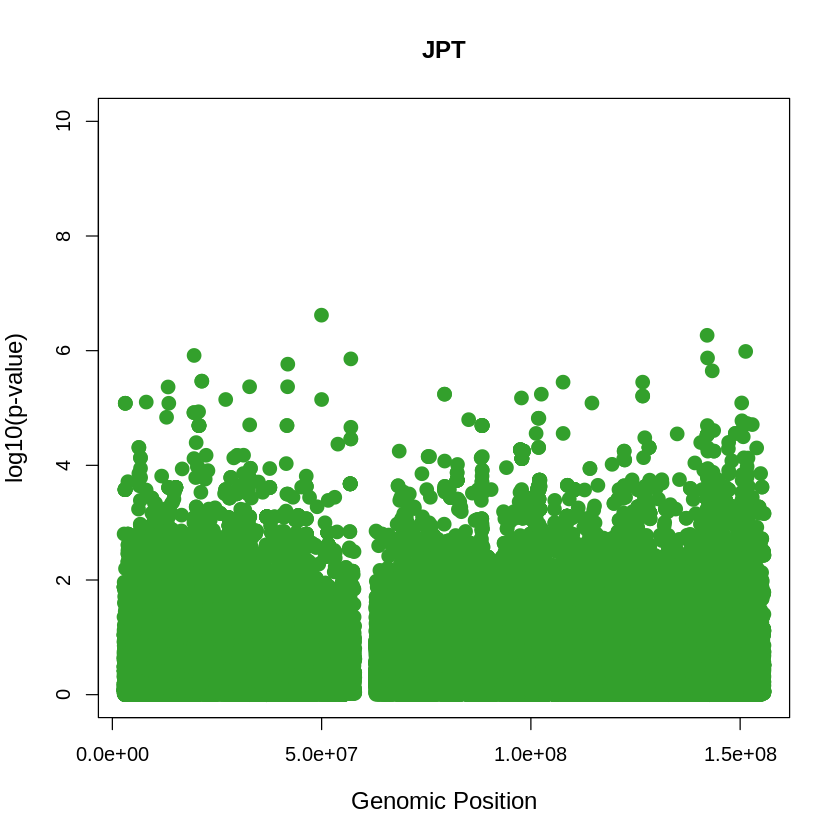
JPT_plot <- plot(JPT$pos, -(JPT$when_mutation_has_freq2), pch = 19, col = "#33A02C", xlab = "Genomic Position", ylab = "log10(p-value)", main = "JPT" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("JPT_plot.pdf", width = 20, height = 5)
JPT_plot <- plot(JPT$pos, -(JPT$when_mutation_has_freq2),
pch = 19, col = "#33A02C",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "JPT",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
KHV <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/KHV/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele", header = TRUE)
head(KHV)| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X257.030640 | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <dbl> | <dbl> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | |
| 1 | 2781635 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.8030350 | -0.6989060 | -0.605342 | -0.45654000 | -0.342867 | -0.503979 | -0.69663300 | 0 | -1.0258700 | -0.847264 |
| 2 | 2781927 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0590766 | -0.0280912 | -0.045688 | -0.00312863 | -0.010792 | -0.018873 | -0.00296262 | 0 | -0.0553768 | -0.635131 |
| 3 | 2781986 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.0590766 | -0.0280912 | -0.045688 | -0.00312863 | -0.010792 | -0.018873 | -0.00296262 | 0 | -0.0553768 | -0.635131 |
| 4 | 2782161 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.9175290 | -1.2623400 | -0.941499 | -1.60266000 | -1.237230 | -1.025680 | -1.11297000 | 0 | -1.2013300 | -0.406086 |
| 5 | 2784102 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.3344400 | -2.3292500 | -1.154890 | -1.79781000 | -1.452120 | -1.538880 | -1.08854000 | 0 | -2.0425200 | -0.471176 |
| 6 | 2784738 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | 1.0000000 | 1.0000000 | 1.000000 | 1.00000000 | 1.000000 | 1.000000 | -0.56223200 | 0 | -0.9722480 | -0.418237 |
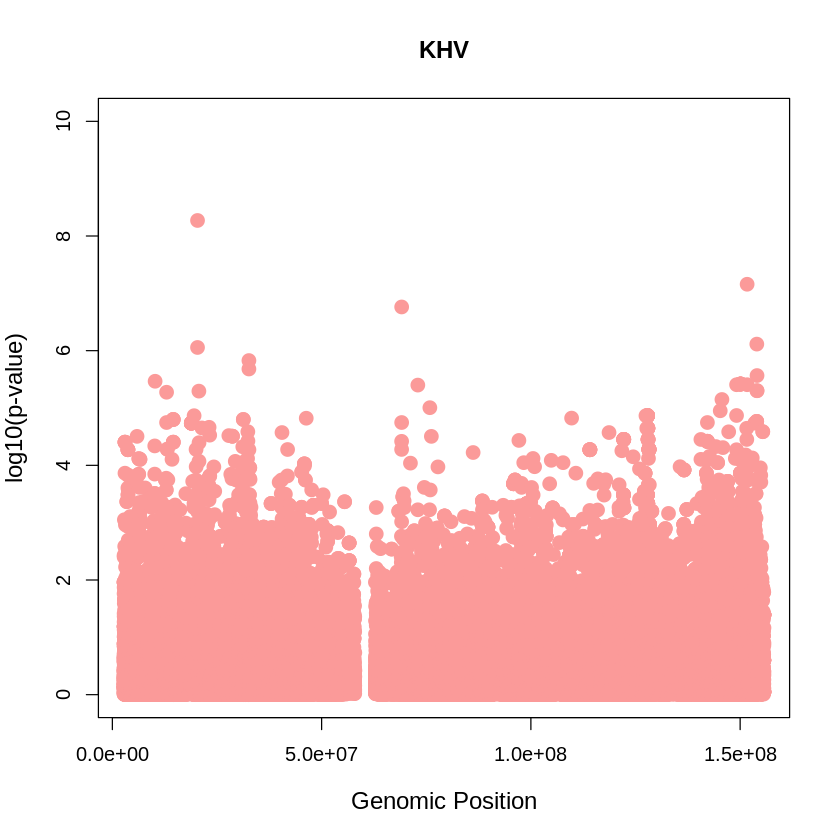
KHV_plot <- plot(KHV$pos, -(KHV$when_mutation_has_freq2), pch = 19, col = "#FB9A99", xlab = "Genomic Position", ylab = "log10(p-value)", main = "KHV" , cex = 1.5, ylim = c(0, 10), cex.lab = 1.2)
pdf("KHV_plot.pdf", width = 20, height = 5)
KHV_plot <- plot(KHV$pos, -(KHV$when_mutation_has_freq2),
pch = 19, col = "#FB9A99",
xlab = "Genomic Position",
ylab = "log10(p-value)",
main = "KHV",
cex = 1.5,
yaxp = c(0, 10, 5),
cex.lab = 1.3,
cex.main = 1.6, # Adjust title size
cex.axis = 1.2) # Adjust axis numbers size
dev.off()
png: 2
library(wesanderson)
# Combine data frames
CDX$Population <- "CDX"
CHB$Population <- "CHB"
CHS$Population <- "CHS"
JPT$Population <- "JPT"
KHV$Population <- "KHV"
combined_data <- rbind(CDX, CHB, CHS, JPT, KHV)
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
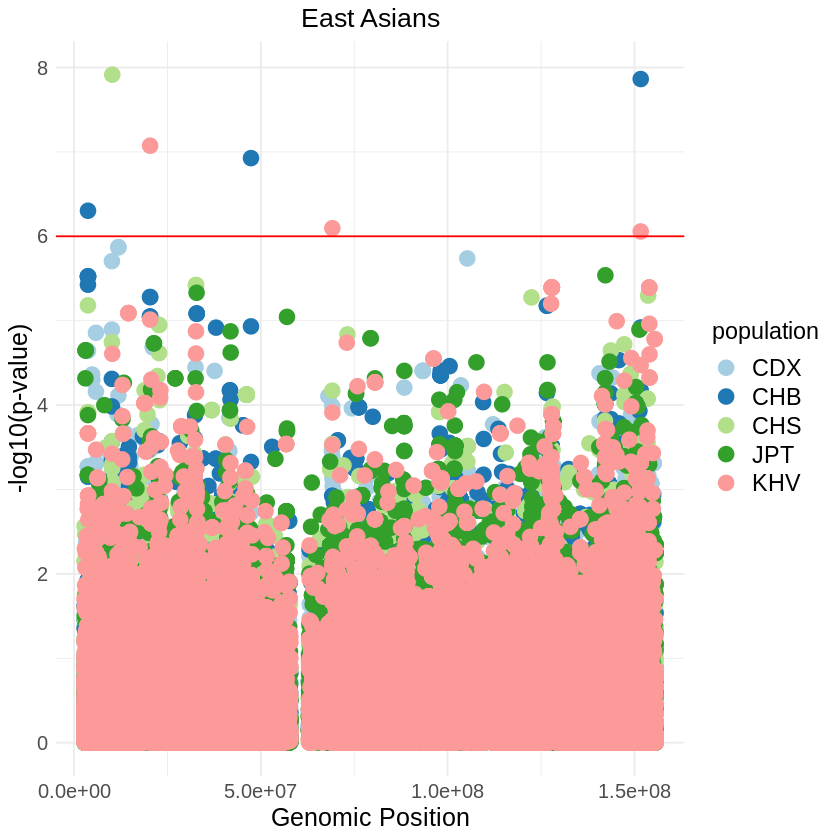
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = -(when_mutation_has_freq2), color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "log10(p-value)", title = "East Asians") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_brewer(palette = "Paired") +
theme(text = element_text(size = 15))
print(plot_combined)
ggsave("asians_threshold_REL_SQUARE.png", plot_combined, width = 7, height = 5)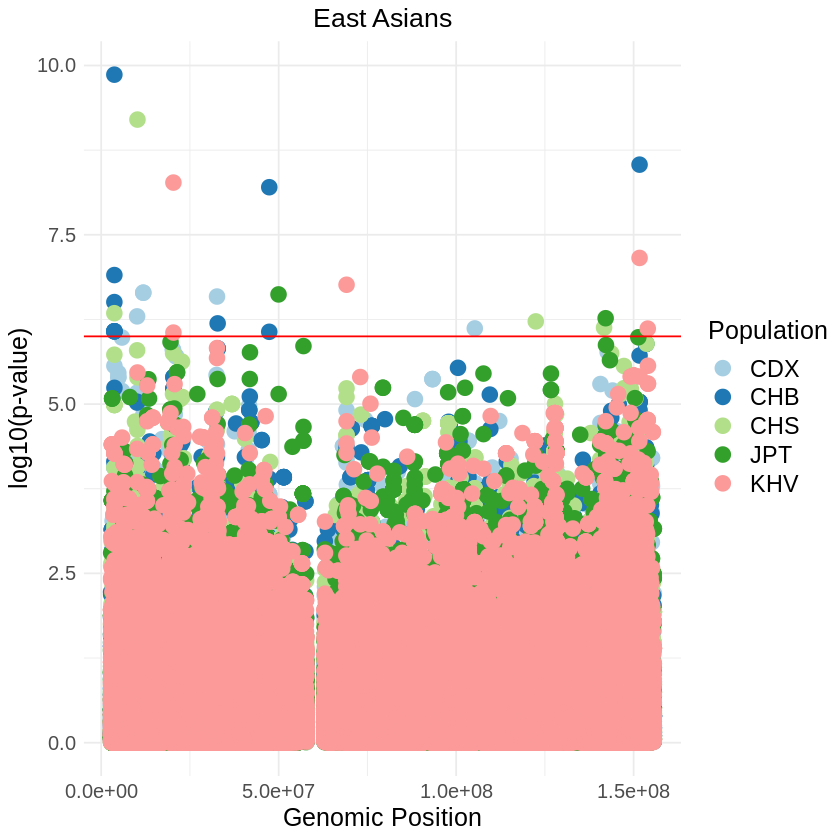
## SELECT JUST POSITION LOWER THAN THRESHOLD (log10 p-value < -6)
# Define file paths for the five .sele files
file_paths_asia <- c(
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/CDX/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/CHB/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/CHS/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/JPT/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/KHV/relate/run_relate/1000g_ppl_phased_haplotypes_selection.sele"
)
# Read the files
CDX <- read.table(file_paths_asia[1], header = TRUE)
CHB <- read.table(file_paths_asia[2], header = TRUE)
CHS <- read.table(file_paths_asia[3], header = TRUE)
JPT <- read.table(file_paths_asia[4], header = TRUE)
KHV <- read.table(file_paths_asia[5], header = TRUE)
# Function to subset data based on log10 p-value
subset_data <- function(data, population) {
subset(data, when_mutation_has_freq2 < -6) %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = CDX, population = "CDX"),
list(data = CHB, population = "CHB"),
list(data = CHS, population = "CHS"),
list(data = JPT, population = "JPT"),
list(data = KHV, population = "KHV")
)
# Subset and bind data from all files
asians_low_pvalue <- do.call(rbind, lapply(file_pop, function(fp) {
subset_data(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(asians_low_pvalue, "asians_low_pvalue.csv", row.names = FALSE)
filtered_asia <- read.csv("/home/ari/ari-intern/people/ari/ariadna-intern/notebooks/asians_low_pvalue.csv")
head(filtered_asia)
cat("Number of rows and columns:", paste(dim(filtered_asia), collapse = "x"))| pos | rs_id | X3571428.500000 | X357142.937500 | X257030.656250 | X184981.281250 | X133128.375000 | X95810.585938 | X68953.507812 | X49624.847656 | ⋯ | X184.981262 | X133.128357 | X95.810577 | X68.953499 | X49.624844 | X35.714287 | X0.000000 | when_DAF_is_half | when_mutation_has_freq2 | population | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <chr> | <int> | <int> | <int> | <int> | <int> | <int> | <int> | <int> | ⋯ | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | <chr> | |
| 1 | 10122953 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.987813 | -0.961790 | -1.191430 | -0.559613 | -0.761888 | -1.644260 | 0 | -1.39061 | -6.29509 | CDX |
| 2 | 11859476 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -2.101280 | -1.576190 | -0.977768 | -1.610050 | -1.664170 | -0.557903 | 0 | -2.69332 | -6.64483 | CDX |
| 3 | 11864438 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -2.101280 | -1.576190 | -0.977768 | -1.610050 | -1.664170 | -0.557903 | 0 | -2.69332 | -6.64483 | CDX |
| 4 | 32635171 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -1.181500 | -0.933064 | -0.809430 | -0.399554 | -0.202704 | -0.298736 | 0 | -2.22411 | -6.58804 | CDX |
| 5 | 105249963 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.612386 | -0.384370 | -0.240353 | -0.152790 | -0.228024 | -0.150998 | 0 | -1.40614 | -6.11750 | CDX |
| 6 | 3712725 | . | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ⋯ | -0.439584 | -0.609713 | -0.366916 | -0.877111 | -0.129806 | 0.000000 | 0 | -1.08669 | -6.50623 | CHB |
Number of rows and columns: 27x36plot_threshold <- ggplot(filtered_asia, aes(x = pos, y = -(when_mutation_has_freq2), color = population)) +
geom_point(size = 4) +
labs(x = "Genomic position", y = "log10 p-value", title = "East Asian Populations") +
theme_minimal() + scale_color_brewer(palette = "Paired") + theme(text = element_text(size = 15),
plot.title = element_text(hjust = 0.5))
print(plot_threshold)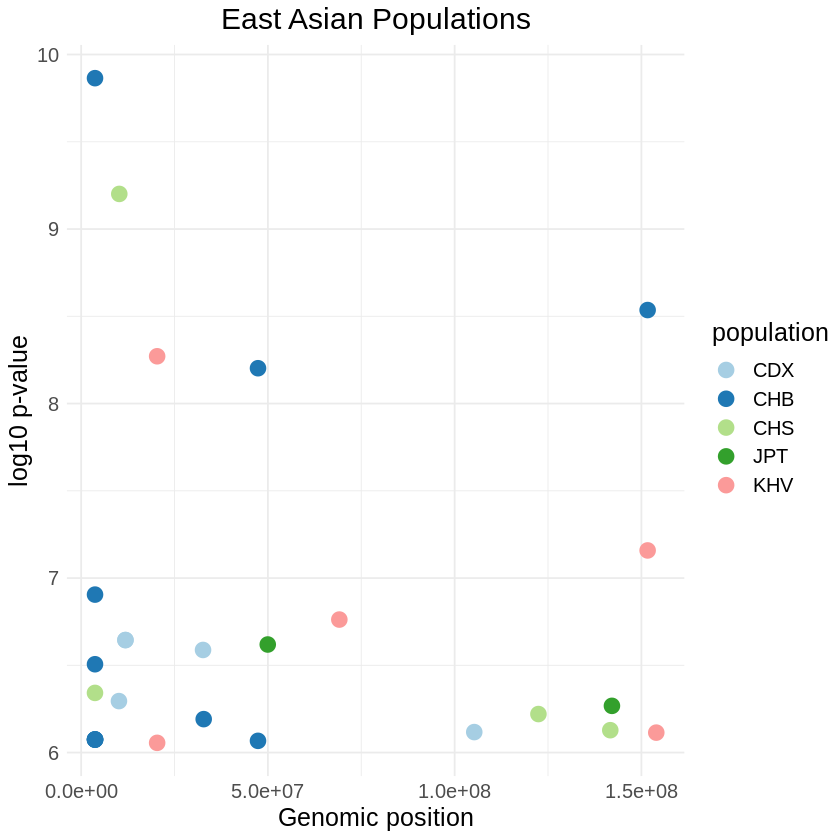
## PLOT TOGETHER ALL POPULATIONS SNPS BELOW THRESHOLD
# Define theme with larger text size for legend
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create plots with the custom theme
africa <- ggplot(filtered, aes(x = pos, y = -(when_mutation_has_freq2), color = population)) +
geom_point(size = 6) +
labs(x = NULL, y = NULL, title = "Africans") +
custom_theme + # Apply the custom theme
scale_color_brewer(palette = "Set2") +
ylim(6, 10)
europe <- ggplot(filtered_euro, aes(x = pos, y = -(when_mutation_has_freq2), color = population)) +
geom_point(size = 6) +
labs(x = NULL, y = NULL, title = "Europeans + PUR") +
custom_theme + # Apply the custom theme
scale_color_manual(values = c("GBR" = "lightblue", "FIN" = "lightpink", "IBS" = "gold", "TSI" = "lightgreen", "PUR" = "orange2")) +
ylim(6, 10)
asia <- ggplot(filtered_asia, aes(x = pos, y = -(when_mutation_has_freq2), color = population)) +
geom_point(size = 6) +
labs(x = NULL, y = NULL, title = "East Asians") +
custom_theme + # Apply the custom theme
scale_color_manual(values = c("CDX" = "#A6CEE3", "CHB" = "#1F78B4", "CHS" = "#B2DF8A", "JPT" = "#33A02C", "KHV" = "#FB9A99")) +
ylim(6, 10)
# Combine the plots
combined_plots <- grid.arrange(africa, europe, asia, ncol = 1,
left = "log10(p-value)",
bottom = "Genomic position")
# Save the combined plot with larger text sizes
ggsave("combined_threshold_RELATE.png", combined_plots, width = 7, height = 15) Warning message:
“Removed 5 rows containing missing values (`geom_point()`).”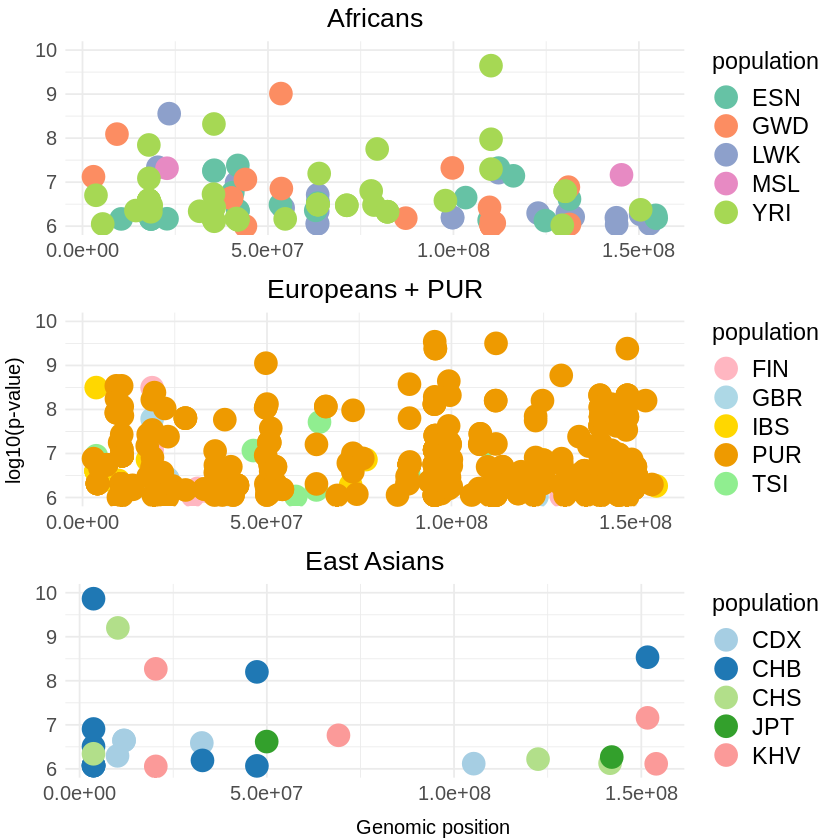
RUNS TEST
AFRICAN POPULATIONS
# Read the CSV files
LWK <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_LWK_runstats.csv", sep=",", header=TRUE)
GWD <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_GWD_runstats.csv", sep=",", header=TRUE)
ESN <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_ESN_runstats.csv", sep=",", header=TRUE)
MSL <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_MSL_runstats.csv", sep=",", header=TRUE)
YRI <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_YRI_runstats.csv", sep=",", header=TRUE)
# Store the datasets in a list
datasets <- list(LWK=LWK, GWD=GWD, ESN=ESN, MSL=MSL, YRI=YRI)
# Loop over each dataset and modify them in place
for (name in names(datasets)) {
# Get the dataset
data <- datasets[[name]]
# Filter the data
filtered_data <- data %>%
filter(stat_name == 'nr_runs' & nr_mut > 20)
# Calculate -log10(p)
filtered_data$log10p <- -log10(filtered_data$p)
# Ensure 'stat' is treated as a factor
filtered_data$stat <- as.factor(filtered_data$stat)
# Assign the modified dataset back to the original variable name
assign(name, filtered_data)
}
# Combine data frames
LWK$Population <- "LWK"
ESN$Population <- "ESN"
GWD$Population <- "GWD"
MSL$Population <- "MSL"
YRI$Population <- "YRI"
combined_data <- rbind(LWK, ESN, GWD, MSL, YRI)
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = log10p, color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "Africans") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_brewer(palette = "Set2") +
theme(
text = element_text(size = 15),
plot.title = element_text(hjust = 0.5) # Center the title
)
# Print the plot
print(plot_combined)
ggsave("africans_threshold_RUNS_SQUARE.png", plot_combined, width = 7, height = 5)ERROR: Error: object 'stat_name' not found
Error: object 'stat_name' not found
Traceback:
1. data %>% filter(stat_name == "nr_runs" & nr_mut > 20)
2. filter(., stat_name == "nr_runs" & nr_mut > 20)# Load necessary libraries
library(dplyr)
library(ggplot2)
# Define the file paths
file_paths <- list(
LWK = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_LWK_runstats.csv",
GWD = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_GWD_runstats.csv",
ESN = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_ESN_runstats.csv",
MSL = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_MSL_runstats.csv",
YRI = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_YRI_runstats.csv"
)
# Read the CSV files
datasets <- lapply(file_paths, function(path) {
read.csv(path, stringsAsFactors = FALSE)
})
# Filter and process each dataset
datasets <- lapply(datasets, function(data) {
data %>%
filter(stat_name == 'nr_runs' & nr_mut > 20) %>%
mutate(
log10p = -log10(p),
stat = as.factor(stat)
)
})
# Combine data frames and add Population column
populations <- names(datasets)
for (i in seq_along(datasets)) {
datasets[[i]]$Population <- populations[i]
}
combined_data <- do.call(rbind, datasets)
# Define the custom theme
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = log10p, color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "Africans") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_brewer(palette = "Set2") +
theme(
text = element_text(size = 15),
plot.title = element_text(hjust = 0.5) # Center the title
)
# Print the plot
print(plot_combined)
ggsave("africans_threshold_RUNS_SQUARE.png", plot_combined, width = 7, height = 5)
Attaching package: ‘dplyr’
The following object is masked from ‘package:gridExtra’:
combine
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Warning message:
“Removed 2353 rows containing missing values (`geom_point()`).”
Warning message:
“Removed 2353 rows containing missing values (`geom_point()`).”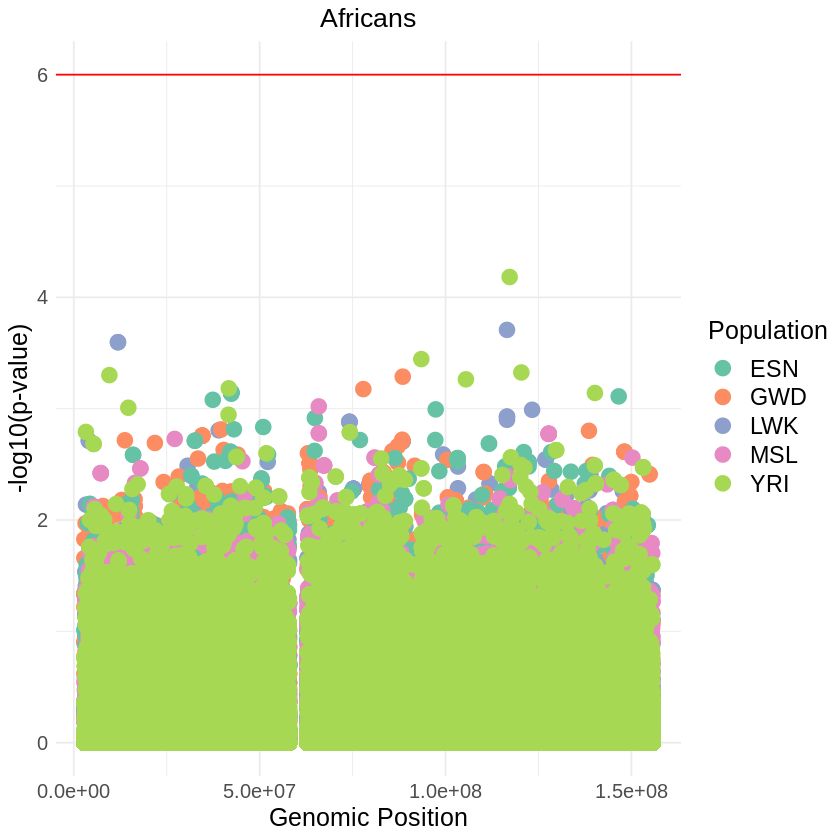
EUROPEANS + PUR
# Read the CSV files
GBR <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_GBR_runstats.csv", sep=",", header=TRUE)
FIN <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_FIN_runstats.csv", sep=",", header=TRUE)
IBS <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_IBS_runstats.csv", sep=",", header=TRUE)
TSI <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_TSI_runstats.csv", sep=",", header=TRUE)
PUR <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_PUR_runstats.csv", sep=",", header=TRUE)
# Store the datasets in a list
datasets <- list(GBR=GBR, FIN=FIN, IBS=IBS, TSI=TSI, PUR=PUR)
# Loop over each dataset and modify them in place
for (name in names(datasets)) {
# Get the dataset
data <- datasets[[name]]
# Filter the data
filtered_data <- data %>%
filter(stat_name == 'nr_runs' & nr_mut > 20)
# Calculate -log10(p)
filtered_data$log10p <- -log10(filtered_data$p)
# Ensure 'stat' is treated as a factor
filtered_data$stat <- as.factor(filtered_data$stat)
# Assign the modified dataset back to the original variable name
assign(name, filtered_data)
}
# Combine data frames
GBR$Population <- "GBR"
FIN$Population <- "FIN"
IBS$Population <- "IBS"
TSI$Population <- "TSI"
PUR$Population <- "PUR"
combined_data <- rbind(GBR, FIN, IBS, TSI, PUR)
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = log10p, color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "Europeans + PUR") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_manual(values = c("GBR" = "lightblue", "FIN" = "lightpink", "IBS" = "gold", "TSI" = "lightgreen", "PUR" = "orange2"))+
theme(
text = element_text(size = 15),
plot.title = element_text(hjust = 0.5) # Center the title
)
# Print the plot
print(plot_combined)
ggsave("europeans_threshold_RUNS_SQUARE.png", plot_combined, width = 7, height = 5)Warning message:
“Removed 7004 rows containing missing values (`geom_point()`).”
Warning message:
“Removed 7004 rows containing missing values (`geom_point()`).”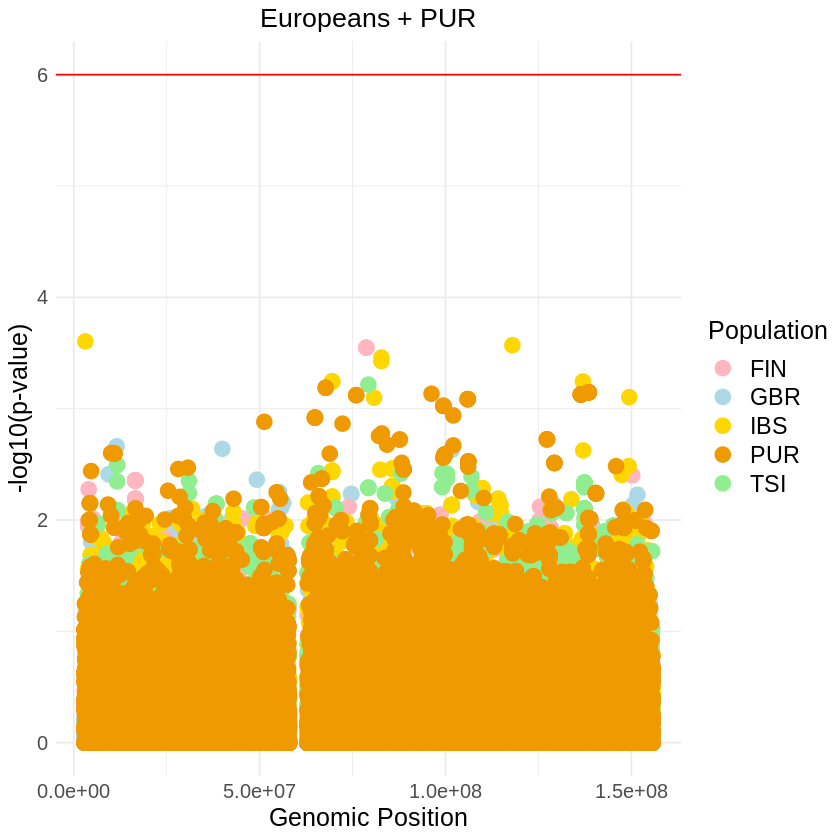
EAST ASIANS
# Load necessary library
library(dplyr)
library(ggplot2)
# Read the CSV files
CDX <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_CDX_runstats.csv", sep=",", header=TRUE)
CHB <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_CHB_runstats.csv", sep=",", header=TRUE)
CHS <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_CHS_runstats.csv", sep=",", header=TRUE)
JPT <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_JPT_runstats.csv", sep=",", header=TRUE)
KHV <- read.table("/home/ari/ari-intern/people/ari/ariadna-intern/steps/runs_test/1000g_ppl_phased_haplotypes_KHV_runstats.csv", sep=",", header=TRUE)
# Store the datasets in a list
datasets <- list(CDX=CDX, CHB=CHB, CHS=CHS, JPT=JPT, KHV=KHV)
# Loop over each dataset and modify them in place
for (name in names(datasets)) {
# Get the dataset
data <- datasets[[name]]
# Filter the data
filtered_data <- data %>%
filter(stat_name == 'nr_runs' & nr_mut > 20)
# Calculate -log10(p)
filtered_data$log10p <- -log10(filtered_data$p)
# Ensure 'stat' is treated as a factor
filtered_data$stat <- as.factor(filtered_data$stat)
# Assign the modified dataset back to the original variable name
assign(name, filtered_data)
}
# Combine data frames
CDX$Population <- "CDX"
CHB$Population <- "CHB"
CHS$Population <- "CHS"
JPT$Population <- "JPT"
KHV$Population <- "KHV"
combined_data <- rbind(CDX, CHB, CHS, JPT, KHV)
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = log10p, color = Population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "East Asians") +
theme_light() + custom_theme +
geom_hline(yintercept = 6, color = "red") +
scale_color_brewer(palette = "Paired") +
theme(
text = element_text(size = 15),
plot.title = element_text(hjust = 0.5) # Center the title
)
# Print the plot
print(plot_combined)
ggsave("asians_threshold_RUNS_SQUARE_correct.png", plot_combined, width = 7, height = 5)Warning message:
“Removed 5769 rows containing missing values (`geom_point()`).”
Warning message:
“Removed 5769 rows containing missing values (`geom_point()`).”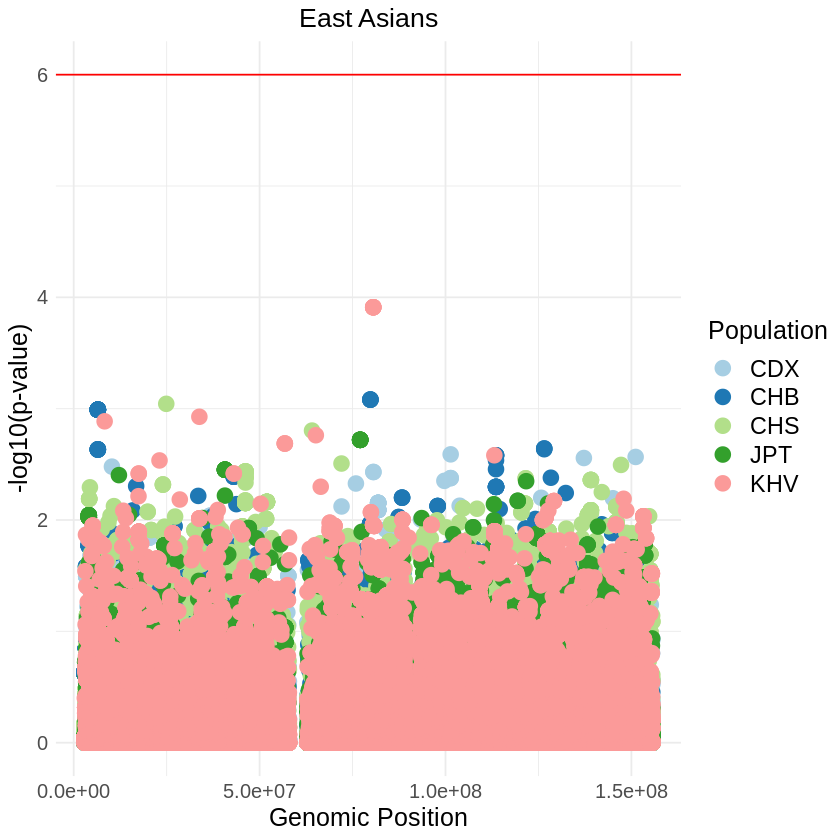
COMBINED P-VALUES
# Load the necessary library
library(dplyr)
# Define file paths for the five .sele files
file_paths_africa <- c(
LWK = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_LWK.csv",
ESN = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_ESN.csv",
GWD = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_GWD.csv",
MSL = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_MSL.csv",
YRI = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_YRI.csv"
)
# Read the files
LWK <- read.csv(file_paths_africa["LWK"], header = TRUE)
ESN <- read.csv(file_paths_africa["ESN"], header = TRUE)
GWD <- read.csv(file_paths_africa["GWD"], header = TRUE)
MSL <- read.csv(file_paths_africa["MSL"], header = TRUE)
YRI <- read.csv(file_paths_africa["YRI"], header = TRUE)
# Function to subset data based on combined_p_values threshold
subset_data <- function(data, population) {
data %>%
filter(combined_p_values >= 6) %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = LWK, population = "LWK"),
list(data = ESN, population = "ESN"),
list(data = GWD, population = "GWD"),
list(data = MSL, population = "MSL"),
list(data = YRI, population = "YRI")
)
# Subset and bind data from all files
africans_high_pvalue <- do.call(rbind, lapply(file_pop, function(fp) {
subset_data(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(africans_high_pvalue, "africans_combined.csv", row.names = FALSE)
Attaching package: ‘dplyr’
The following object is masked from ‘package:gridExtra’:
combine
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
library(dplyr)
## SELECT JUST POSITION LOWER THAN THRESHOLD (log10 p-value < -6)
# Define file paths for the five .sele files
file_paths_euro <- c(
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_GBR.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_FIN.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_IBS.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_TSI.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_PUR.csv"
)
# Read the files
GBR <- read.csv(file_paths_euro[1], header = TRUE)
FIN <- read.csv(file_paths_euro[2], header = TRUE)
IBS <- read.csv(file_paths_euro[3], header = TRUE)
TSI <- read.csv(file_paths_euro[4], header = TRUE)
PUR <- read.csv(file_paths_euro[5], header = TRUE)
# Function to subset data based on combined_p_values threshold
subset_data <- function(data, population) {
data %>%
filter(combined_p_values >= 6) %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = GBR, population = "GBR"),
list(data = FIN, population = "FIN"),
list(data = IBS, population = "IBS"),
list(data = TSI, population = "TSI"),
list(data = PUR, population = "PUR")
)
# Subset and bind data from all files
europeans_low_pvalue <- do.call(rbind, lapply(file_pop, function(fp) {
subset_data(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(europeans_low_pvalue, "europeans_combined.csv", row.names = FALSE)# Define file paths for the five .csv files
file_paths_asia_combined <- c(
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_CDX.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_CHB.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_CHS.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_JPT.csv",
"/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_KHV.csv"
)
# Read the files
CDX <- read.csv(file_paths_asia_combined[1])
CHB <- read.csv(file_paths_asia_combined[2])
CHS <- read.csv(file_paths_asia_combined[3])
JPT <- read.csv(file_paths_asia_combined[4])
KHV <- read.csv(file_paths_asia_combined[5])
# Add population column to each data frame
CDX$population <- "CDX"
CHB$population <- "CHB"
CHS$population <- "CHS"
JPT$population <- "JPT"
KHV$population <- "KHV"
# Function to subset data based on combined_p_values
subset_data <- function(data, population) {
data %>%
filter(combined_p_values >= 6) %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = CDX, population = "CDX"),
list(data = CHB, population = "CHB"),
list(data = CHS, population = "CHS"),
list(data = JPT, population = "JPT"),
list(data = KHV, population = "KHV")
)
# Subset and bind data from all files
asians_combined <- do.call(rbind, lapply(file_pop, function(fp) {
subset_data(fp$data, fp$population)
}))
# Save the combined data to a single CSV file
write.csv(asians_combined, "asians_combined.csv", row.names = FALSE)africa_file <- read.csv("/home/ari/ari-intern/people/ari/ariadna-intern/notebooks/africans_combined.csv")
europe_file <- read.csv("/home/ari/ari-intern/people/ari/ariadna-intern/notebooks/europeans_combined.csv")
asia_file <- read.csv("/home/ari/ari-intern/people/ari/ariadna-intern/notebooks/asians_combined.csv")
# Define theme with larger text size for legend
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create plots with the custom theme
africa <- ggplot(africa_file, aes(x = pos, y = (combined_p_values), color = population)) +
geom_point(size = 6) +
labs(x = NULL, y = NULL, title = "Africans") +
custom_theme + # Apply the custom theme
scale_color_brewer(palette = "Set2") +
ylim(6, 10)
europe <- ggplot(europe_file, aes(x = pos, y = (combined_p_values), color = population)) +
geom_point(size = 6) +
labs(x = NULL, y = NULL, title = "Europeans + PUR") +
custom_theme + # Apply the custom theme
scale_color_manual(values = c("GBR" = "lightblue", "FIN" = "lightpink", "IBS" = "gold", "TSI" = "lightgreen", "PUR" = "orange2")) +
ylim(6, 10)
asia <- ggplot(asia_file, aes(x = pos, y = (combined_p_values), color = population)) +
geom_point(size = 6) +
labs(x = NULL, y = NULL, title = "East Asians") +
custom_theme + # Apply the custom theme
scale_color_manual(values = c("CDX" = "#A6CEE3", "CHB" = "#1F78B4", "CHS" = "#B2DF8A", "JPT" = "#33A02C", "KHV" = "#FB9A99")) +
ylim(6, 10)
# Combine the plots
combined_plots <- grid.arrange(africa, europe, asia, ncol = 1,
left = "log10(p-value)",
bottom = "Genomic position")
# Save the combined plot with larger text sizes
ggsave("combined_threshold_COMBINED_gap.png", combined_plots, width = 7, height = 15) Warning message:
“Removed 3 rows containing missing values (`geom_point()`).”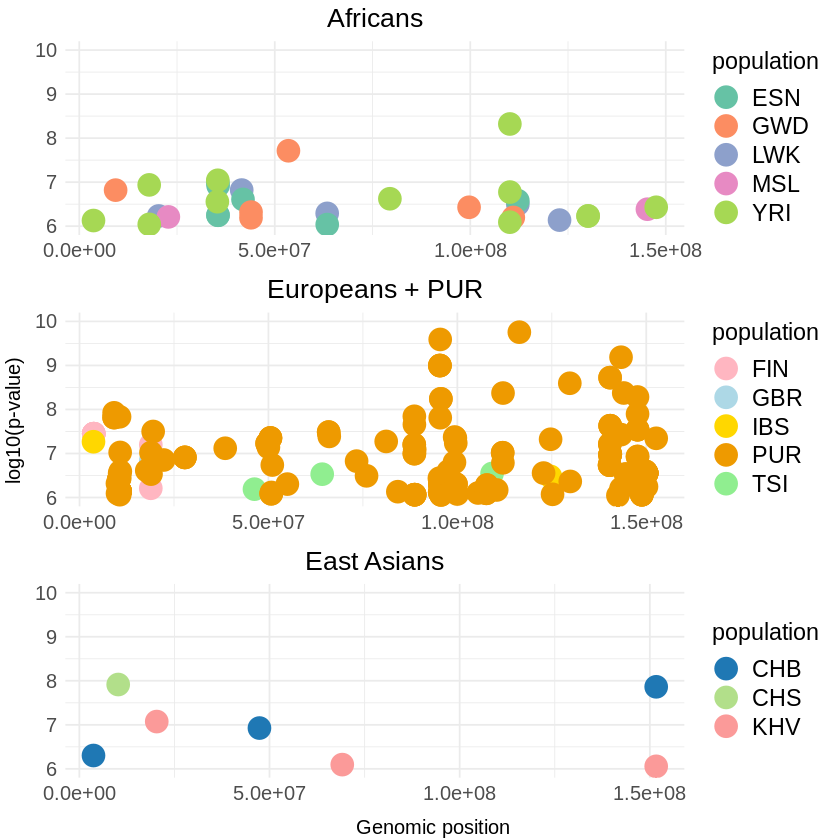
# Load the necessary libraries
library(dplyr)
library(ggplot2)
# Define file paths for the five .sele files
file_paths_africa <- c(
LWK = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_LWK.csv",
ESN = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_ESN.csv",
GWD = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_GWD.csv",
MSL = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_MSL.csv",
YRI = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_YRI.csv"
)
# Read the files
LWK <- read.csv(file_paths_africa["LWK"], header = TRUE)
ESN <- read.csv(file_paths_africa["ESN"], header = TRUE)
GWD <- read.csv(file_paths_africa["GWD"], header = TRUE)
MSL <- read.csv(file_paths_africa["MSL"], header = TRUE)
YRI <- read.csv(file_paths_africa["YRI"], header = TRUE)
# Function to add population column to the data
add_population <- function(data, population) {
data %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = LWK, population = "LWK"),
list(data = ESN, population = "ESN"),
list(data = GWD, population = "GWD"),
list(data = MSL, population = "MSL"),
list(data = YRI, population = "YRI")
)
# Add population column and bind data from all files
africans_combined <- do.call(rbind, lapply(file_pop, function(fp) {
add_population(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(africans_combined, "africans_combined.csv", row.names = FALSE)
# Define theme with larger text size for legend
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = africans_combined, aes(x = pos, y = combined_p_values, color = population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "Africans") +
theme_light() +
custom_theme +
scale_color_brewer(palette = "Set2") +
geom_hline(yintercept = 6, color = "red")
# Print the plot
print(plot_combined)
ggsave("africans_combined_pvalues_SQUARE_big.png", plot_combined, width = 7, height = 5)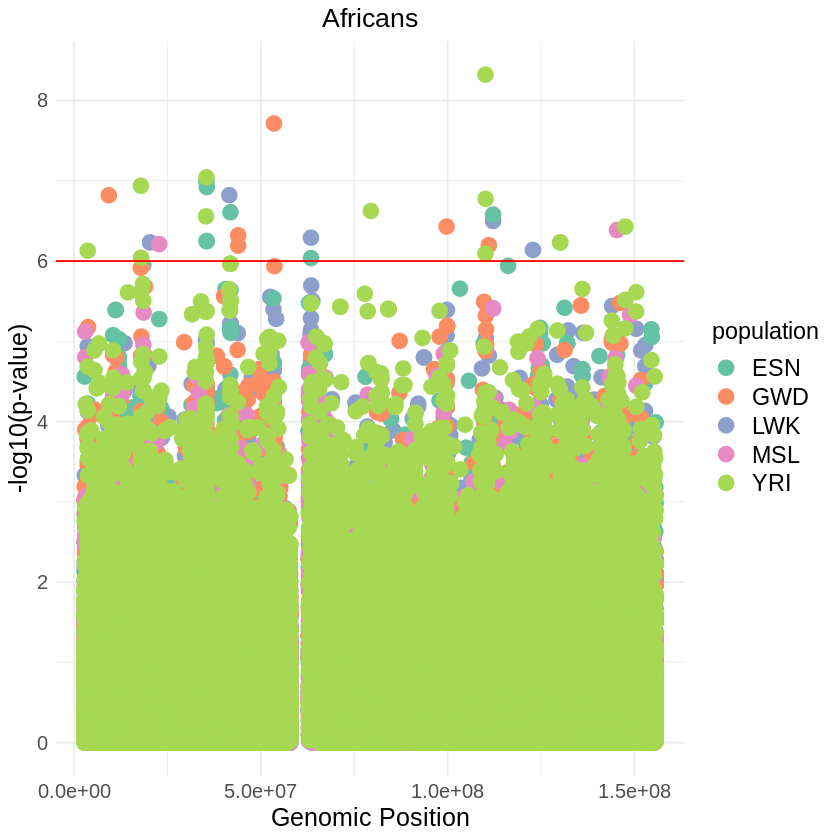
# Load the necessary libraries
library(dplyr)
library(ggplot2)
# Define file paths for the five .sele files
file_paths <- c(
GBR = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_GBR.csv",
FIN = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_FIN.csv",
IBS = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_IBS.csv",
TSI = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_TSI.csv",
PUR = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_PUR.csv"
)
# Read the files
GBR <- read.csv(file_paths["GBR"], header = TRUE)
FIN <- read.csv(file_paths["FIN"], header = TRUE)
IBS <- read.csv(file_paths["IBS"], header = TRUE)
TSI <- read.csv(file_paths["TSI"], header = TRUE)
PUR <- read.csv(file_paths["PUR"], header = TRUE)
# Function to add population column to the data
add_population <- function(data, population) {
data %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = GBR, population = "GBR"),
list(data = FIN, population = "FIN"),
list(data = IBS, population = "IBS"),
list(data = TSI, population = "TSI"),
list(data = PUR, population = "PUR")
)
# Add population column and bind data from all files
combined_data <- do.call(rbind, lapply(file_pop, function(fp) {
add_population(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(combined_data, "european_populations.csv", row.names = FALSE)
# Create the plot
plot_combined <- ggplot(data = combined_data, aes(x = pos, y = combined_p_values, color = population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "Europeans+PUR") +
theme_light() +
custom_theme+
scale_color_manual(values = c(
"GBR" = "lightblue",
"FIN" = "lightpink",
"IBS" = "gold",
"TSI" = "lightgreen",
"PUR" = "orange2"
)) +
geom_hline(yintercept = 6, color = "red")
# Print the plot
print(plot_combined)
ggsave("europeans_combined_pvalues_SQUARE_big.png", plot_combined, width = 7, height = 5)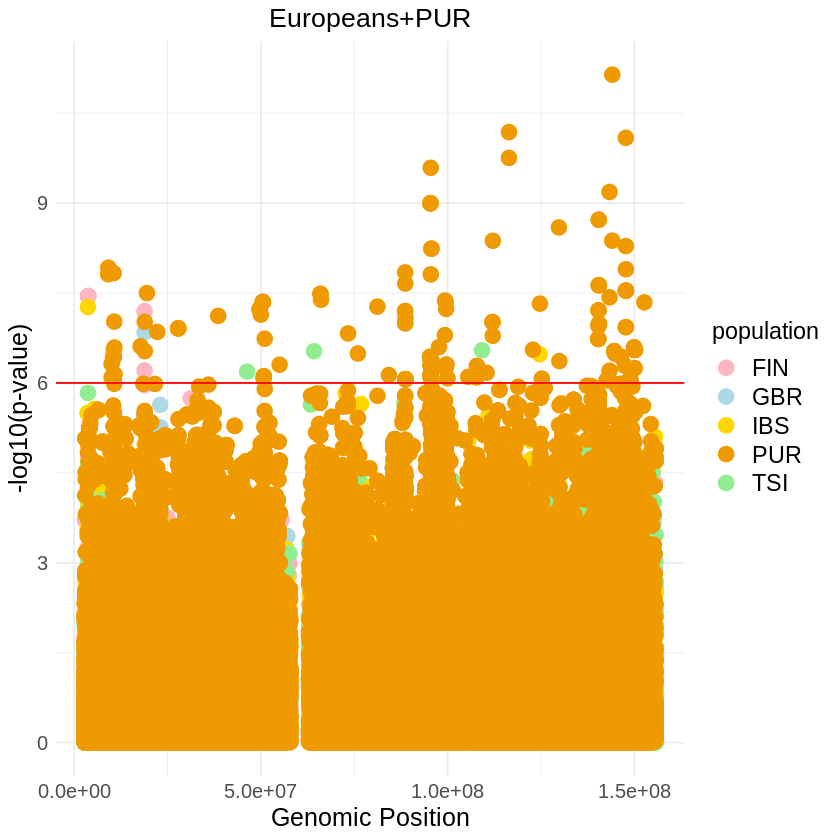
# Define file paths for the five .sele files
file_paths_asia <- c(
CDX = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_CDX.csv",
CHB = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_CHB.csv",
CHS = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_CHS.csv",
JPT = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_JPT.csv",
KHV = "/home/ari/ari-intern/people/ari/ariadna-intern/steps/fisher/combined_KHV.csv"
)
# Read the files
CDX <- read.csv(file_paths_asia["CDX"], header = TRUE)
CHB <- read.csv(file_paths_asia["CHB"], header = TRUE)
CHS <- read.csv(file_paths_asia["CHS"], header = TRUE)
JPT <- read.csv(file_paths_asia["JPT"], header = TRUE)
KHV <- read.csv(file_paths_asia["KHV"], header = TRUE)
# Function to add population column to the data
add_population <- function(data, population) {
data %>%
mutate(population = population)
}
# Define data frames and corresponding population names
file_pop <- list(
list(data = CDX, population = "CDX"),
list(data = CHB, population = "CHB"),
list(data = CHS, population = "CHS"),
list(data = JPT, population = "JPT"),
list(data = KHV, population = "KHV")
)
# Add population column and bind data from all files
asians_combined <- do.call(rbind, lapply(file_pop, function(fp) {
add_population(fp$data, fp$population)
}))
# Save the combined data to a single file
write.csv(asians_combined, "asians_combined.csv", row.names = FALSE)
# Define theme with larger text size for legend
custom_theme <- theme_minimal() +
theme(
text = element_text(size = 14), # Increase the text size for all text elements
plot.title = element_text(size = 16, hjust = 0.5), # Increase the title size
axis.title = element_text(size = 15), # Increase the axis label size
axis.text = element_text(size = 12), # Increase the axis text size
legend.text = element_text(size = 14) # Increase the legend text size
)
# Create the plot
plot_combined <- ggplot(data = asians_combined, aes(x = pos, y = combined_p_values, color = population)) +
geom_point(pch = 19, size = 4) +
labs(x = "Genomic Position", y = "-log10(p-value)", title = "East Asians") +
theme_light() +
custom_theme +
scale_color_brewer(palette = "Paired") +
geom_hline(yintercept = 6, color = "red")
# Print the plot
print(plot_combined)
ggsave("asians_combined_pvalues_SQUARE_big.png", plot_combined, width = 7, height = 5)