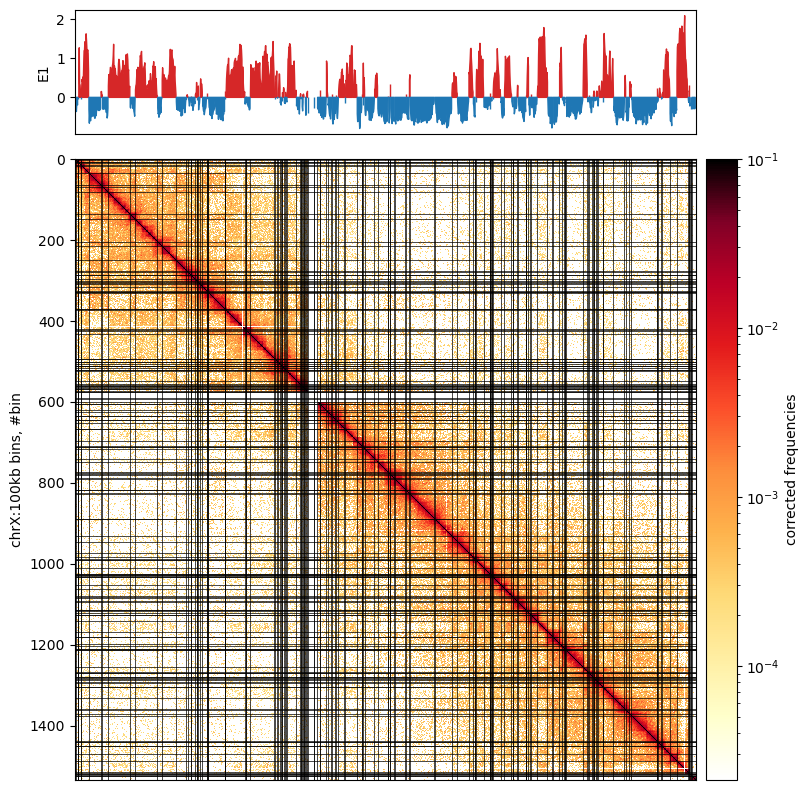
0 1
1 0
2 0
3 1
4 1
..
1529 1
1530 1
1531 1
1532 1
1533 1
Name: E1, Length: 1534, dtype: int64
[-1 0 1 ... 0 0 0]
(array([ 0, 2, 7, 8, 9, 15, 16, 34, 64, 68, 82,
136, 149, 204, 215, 250, 276, 280, 286, 287, 297, 302,
303, 305, 309, 315, 326, 327, 329, 330, 370, 371, 372,
420, 423, 424, 433, 493, 494, 500, 502, 507, 510, 511,
512, 517, 520, 522, 524, 547, 556, 558, 560, 561, 564,
566, 567, 569, 572, 575, 576, 591, 592, 599, 605, 606,
621, 625, 634, 635, 644, 645, 651, 666, 667, 697, 709,
710, 711, 716, 740, 748, 774, 775, 779, 780, 789, 790,
817, 818, 826, 828, 888, 889, 931, 945, 973, 984, 989,
990, 991, 1010, 1025, 1026, 1027, 1028, 1031, 1032, 1033, 1052,
1061, 1079, 1083, 1084, 1092, 1095, 1114, 1115, 1116, 1121, 1126,
1127, 1142, 1168, 1180, 1181, 1197, 1204, 1210, 1211, 1212, 1213,
1255, 1266, 1269, 1270, 1279, 1280, 1282, 1284, 1288, 1291, 1294,
1304, 1305, 1331, 1358, 1361, 1370, 1375, 1438, 1439, 1441, 1449,
1451, 1470, 1487, 1515, 1517, 1518, 1520, 1522, 1523, 1524]),)