# import standard python libraries
import numpy as np
import pandas as pd
# Import packages for working with cooler files and tools for plots
import cooler
import cooltools.lib.plotting
import cooltools
import seaborn as sns
from cooltools.lib.numutils import adaptive_coarsegrain, interp_nan
from matplotlib.colors import LogNorm
from mpl_toolkits.axes_grid1 import make_axes_locatable
Various Comparisons
Various plots using homemade plotting utility comparing different parameters and methods for the manuscript
Notes before we start
We want to create a plotting utility so we can easily plot and compare different matrices. We might move them to hicstuff.py for import, but as for now, they are defined only in this notebook.
Setup
Imports for plots etc
InlineBackend.rc
For consistent plots that fit with pdf manus.
import matplotlib.pyplot as plt
# Define my params:
## Params will comply with my desired layout for the Manuscript (PDF)
notebook_rcparams = {
'font.size': 7,
'axes.titlesize': 8,
'axes.labelsize': 7,
'xtick.labelsize': 6,
'ytick.labelsize': 6,
'figure.titlesize': 9,
'figure.figsize': [6.0, 2.0],
'figure.labelsize': 7,
}
# Apply config
%matplotlib inline
%config InlineBackend.figure_formats = ['retina']
%config InlineBackend.rc = notebook_rcparamsprint({key:plt.rcParams[key] for key in notebook_rcparams.keys()}){'font.size': 10.0, 'axes.titlesize': 'large', 'axes.labelsize': 'medium', 'xtick.labelsize': 'medium', 'ytick.labelsize': 'medium', 'figure.titlesize': 'large', 'figure.figsize': [6.4, 4.8], 'figure.labelsize': 'large'}## Apparently IPython needs this twice to understand??
import matplotlib.pyplot as plt
# Define my params:
## Params will comply with my desired layout for the Manuscript (PDF)
notebook_rcparams = {
'font.size': 7,
'axes.titlesize': 8,
'axes.labelsize': 7,
'xtick.labelsize': 6,
'ytick.labelsize': 6,
'figure.titlesize': 9,
'figure.figsize': [6.0, 2.0],
'figure.labelsize': 7,
}
# Apply config
%matplotlib inline
%config InlineBackend.figure_formats = ['retina']
%config InlineBackend.rc = notebook_rcparamsprint({key:plt.rcParams[key] for key in notebook_rcparams.keys()}){'font.size': 7.0, 'axes.titlesize': 8.0, 'axes.labelsize': 7.0, 'xtick.labelsize': 6.0, 'ytick.labelsize': 6.0, 'figure.titlesize': 9.0, 'figure.figsize': [6.0, 2.0], 'figure.labelsize': 7.0}Load all the coolers and eigenvectors
## Loading all coolers and their compartments
import glob
import os.path as op
import cooler
# Abbreviate the names (almost the same abbreviations as Wang et al. 2019)
names_abbr = {'fibroblast': 'Fib', 'spermatogonia': 'SPA', 'pachytene_spermatocyte': 'PAC', 'round_spermatid': 'RS', 'sperm': 'Sp'}
# Paths and resolutions
mcools = pd.Series(glob.glob("../steps/bwa/*/cool/*/*.mcool"))
res_100 = "::resolutions/100000"
res_500 = "::resolutions/500000"
# Create a DataFrame
clr_df = pd.DataFrame({
'name': pd.concat([mcools.apply(lambda x: op.basename(op.dirname(x)))] * 3, ignore_index=True),
'resolution': ['100'] * len(mcools) + ['ps500'] * len(mcools) + ['500'] * len(mcools),
'mcool_path': pd.concat([mcools] * 3, ignore_index=True)
})
# Insert the 'sname' column (shortname/abbreviation)
clr_df['sname'] = clr_df['name'].apply(lambda x: names_abbr[x])
# Create the 'cooler_id' column
clr_df['cooler_id'] = clr_df.apply(
lambda row: cooler.Cooler(
row['mcool_path'] + (res_100 if row['resolution'] in ['100', 'ps500'] else res_500)
),
axis=1
)
# Determine parsing run (dirname 3 levels up PE or recPE)
clr_df['run'] = clr_df['mcool_path'].apply(lambda x: 'recPE' if 'rec' in x else 'PE')
# Add the 'nbins' column
clr_df['nbins'] = clr_df['cooler_id'].apply(lambda x: len(x.bins().fetch('chrX')))
# Add the path to the compartments (../results/{compartments,rec_compartments})
# make a comp_dir dict
comp_dirs = {
'PE': '../results/compartments/',
'recPE': '../results/rec_compartments/'
}
clr_df['e1_dict'] = clr_df.apply(
lambda row: {view: pd.read_csv(op.join(
comp_dirs[row['run']], f"{row['name']}_e1_{row['resolution']}kb_{view}.csv"
))['e1']
if row['resolution'] != 'ps500'
else pd.read_csv(op.join(
comp_dirs[row['run']], f"{row['name']}_e1_100kb_{view}_smoothed.csv"
))['e1']
for view in ['full', 'arms', '10Mb']},
axis=1
)
# Now we have all the coolers and compartments ready for plotting (in semi-long format)
#clr_df
clr_df[['sname', 'resolution', 'nbins', 'run', 'e1_dict']].query('resolution == "ps500"')
#print(clr_df['e1_dict'].values)| sname | resolution | nbins | run | e1_dict | |
|---|---|---|---|---|---|
| 10 | PAC | ps500 | 1534 | recPE | {'full': [nan, nan, nan, nan, nan, nan, nan, n... |
| 11 | SPA | ps500 | 1534 | recPE | {'full': [nan, nan, nan, nan, nan, nan, nan, n... |
| 12 | Fib | ps500 | 1534 | recPE | {'full': [nan, nan, nan, nan, nan, -0.15077432... |
| 13 | RS | ps500 | 1534 | recPE | {'full': [nan, nan, nan, nan, nan, nan, nan, n... |
| 14 | Sp | ps500 | 1534 | recPE | {'full': [nan, nan, nan, nan, nan, -0.00698684... |
| 15 | RS | ps500 | 1534 | PE | {'full': [nan, nan, nan, nan, nan, -0.17383142... |
| 16 | SPA | ps500 | 1534 | PE | {'full': [nan, nan, nan, nan, nan, nan, nan, -... |
| 17 | Sp | ps500 | 1534 | PE | {'full': [nan, nan, nan, -0.102536873416233, -... |
| 18 | Fib | ps500 | 1534 | PE | {'full': [nan, nan, nan, -0.2342462339281897, ... |
| 19 | PAC | ps500 | 1534 | PE | {'full': [nan, nan, nan, nan, nan, 0.595328434... |
Load the genomic intervals
A-compartments
import pandas as pd
import os
# ECH regions and chromsizes
ech90 = pd.read_csv('../data/ech90_human_Mmul_10.csv').assign(length=lambda x: x.end - x.start)
chromsizes = (pd.read_csv(
'../data/rheMac10.filtered.chrom.sizes',
sep='\t',
index_col='chrom',
header=None,
names=['chrom','size'])
.to_dict()['size']
)
# Directory containing your .csv files
comp_dirs = {
'PE': '../results/compartments/',
'recPE': '../results/rec_compartments/'
}
# Create a dictionary to store the comps
comps = {
'PE': {},
'recPE': {}
}
# Iterate over all .csv files in the directory
for run in comps.keys():
comp_dir = comp_dirs[run]
for filename in os.listdir(comp_dir):
# We only want the .csv files and not e1 tracks
if filename.endswith('.csv') and 'e1' not in filename:
# Construct the full file path
filepath = os.path.join(comp_dir, filename)
# Load the CSV into a DataFrame
# Use the filename (without extension) as the dictionary key
key = filename.replace('_a_comp_coords_', '_')
key = os.path.splitext(key)[0]
comps[run][key] = pd.read_csv(filepath).assign(length=lambda x: x.end - x.start)
# The `comps` dictionary now contains the DataFrames
# Example usage:
comps['recPE']['round_spermatid_100kb_arms']| chrom | start | end | resolution | length | |
|---|---|---|---|---|---|
| 0 | chrX | 900000 | 1600000 | 100000 | 700000 |
| 1 | chrX | 1700000 | 2600000 | 100000 | 900000 |
| 2 | chrX | 2900000 | 3100000 | 100000 | 200000 |
| 3 | chrX | 3200000 | 3500000 | 100000 | 300000 |
| 4 | chrX | 6600000 | 6700000 | 100000 | 100000 |
| ... | ... | ... | ... | ... | ... |
| 72 | chrX | 145400000 | 147100000 | 100000 | 1700000 |
| 73 | chrX | 147300000 | 147400000 | 100000 | 100000 |
| 74 | chrX | 148800000 | 149100000 | 100000 | 300000 |
| 75 | chrX | 149300000 | 151200000 | 100000 | 1900000 |
| 76 | chrX | 152500000 | 153300000 | 100000 | 800000 |
77 rows × 5 columns
Edge intervals
import pandas as pd
import os
# Directory containing your .csv files
edge_dirs = {
'PE': '../results/edges/',
'recPE': '../results/rec_edges/'
}
edges = {
'PE': {},
'recPE': {}
}
# Iterate over all .csv files in the directories
for run in edges.keys():
edge_dir = edge_dirs[run]
for filename in os.listdir(edge_dir):
if filename.endswith('.csv'): # Check for .csv files
# Construct the full file path
filepath = os.path.join(edge_dir, filename)
# Load the CSV into a DataFrame
# Use the filename (without extension) as the dictionary key
key = filename.replace('_edges', '')
key = os.path.splitext(key)[0]
edges[run][key] = pd.read_csv(filepath).assign(length=lambda x: x.end - x.start)
# The `edges` dictionary now contains the DataFrames
print(edges.keys())
display(edges['recPE']['fibroblast_100kb_10Mb'])
#ech90 = pd.read_csv('../data/ech90_human_Mmul_10.csv')dict_keys(['PE', 'recPE'])| start | end | chrom | resolution | length | |
|---|---|---|---|---|---|
| 0 | 700000 | 900000 | chrX | 100000 | 200000 |
| 1 | 3300000 | 3500000 | chrX | 100000 | 200000 |
| 2 | 8200000 | 8400000 | chrX | 100000 | 200000 |
| 3 | 13300000 | 13500000 | chrX | 100000 | 200000 |
| 4 | 14900000 | 15100000 | chrX | 100000 | 200000 |
| ... | ... | ... | ... | ... | ... |
| 87 | 144400000 | 144800000 | chrX | 100000 | 400000 |
| 88 | 145400000 | 145600000 | chrX | 100000 | 200000 |
| 89 | 146700000 | 147100000 | chrX | 100000 | 400000 |
| 90 | 148800000 | 149000000 | chrX | 100000 | 200000 |
| 91 | 152100000 | 152300000 | chrX | 100000 | 200000 |
92 rows × 5 columns
Baboons
# Load baboons
import pandas as pd
from genominterv import interval_collapse
hama_panu3 = pd.read_csv('../data/lift/papanu4/high_hama_papanu4.bed', sep='\t', index_col=False, header=None, names=['group','chrom', 'start', 'end'])
olive_panu3 = pd.read_csv('../data/lift/papanu4/high_olive_papanu4.bed', sep='\t', index_col=False, header=None, names=['group','chrom', 'start', 'end'])
baboon_df = pd.read_csv('../results/high_baboon_rhemac10.bed', sep='\t', index_col=False, header=None, names=['chrom', 'start', 'end', 'group', 'score'])
high_hama_ucsc = (interval_collapse(baboon_df.query('group == "high_hama"')).
assign(group = "high_hama").
query('chrom == "chrX"')
)
high_olive_ucsc = (interval_collapse(baboon_df.query('group == "high_olive"')).
assign(group = "high_olive").
query('chrom == "chrX"')
)
high_hama = pd.read_csv('../data/lift/rheMac10/high_hama_rhemac10.bed', sep='\t', index_col=False, header=None, names=['group','chrom', 'start', 'end'])
high_olive = pd.read_csv('../data/lift/rheMac10/high_olive_rhemac10.bed', sep='\t', index_col=False, header=None, names=['group','chrom', 'start', 'end'])
baboon_dict = {"P.hama": high_hama, "P.anubis": high_olive}Define a plotting utility for compartments and E1
This plots a group/subset of coolers and their respective E1 compartments.
# We can vary the number of subplots based on the number of coolers
# We can vary the number of views for each cooler
# We can define the region (tuple) of the chromosome to plot (default chrX)
def plot_grouped(
group: pd.DataFrame,
region: str = 'chrX', # or (chrom, start, end)
include_compartments: bool = True,
view_order: list[str] = ['10Mb', 'arms', 'full'],
figsize: tuple[float, float] = (6, 6)
) -> None:
group = group.reset_index(drop=True)
# Define the number of subplots based on the number of coolers
f, axs = plt.subplots(1,group.shape[0],
figsize=figsize,
sharex=True
)
# Logtransform the colorscale
norm = LogNorm(vmax=0.1)
# If there is only one cooler, axs is not a list, so we need to make it a list
if not isinstance(axs, np.ndarray):
axs = [axs]
# Loop through the clrs and its matrix: plot the matrix on its axis
for row in group.itertuples():
ax = axs[row.Index]
# Define the y-axis label
title = f'{row.sname}{row.resolution}kb: {row.run}'
ylab = f'{region}:{row.resolution}kb [Mb]'
# Extract some variables
e1_dict = {k: v for k, v in row.e1_dict.items() if k in view_order}
chrom_start = row.cooler_id.bins().fetch(region)['start'].values
chrom_end = row.cooler_id.bins().fetch(region)['end'].values
im = ax.matshow(
row.cooler_id.matrix().fetch(region),
norm=norm,
cmap='fall',
);
ax.set_xlim(0, row.nbins)
ax.set_ylim(row.nbins, 0)
ax.set_ylabel(ylab)
ticks = np.linspace(0, row.nbins, 5)
ax.set_yticks(ticks)
ax.set_yticklabels([f'{x*row.cooler_id.binsize/1_000_000:.0f}' for x in ticks])
ax.xaxis.set_visible(False)
divider = make_axes_locatable(ax)
cax = divider.append_axes("right", size="2%", pad=0.05)
plt.colorbar(im, cax=cax, label='corrected frequencies');
# Only show the colorbar on the last plot and ytitle on first plot
if row.Index != group.shape[0] - 1:
cax.set_visible(False)
elif row.Index != 0:
ax.set_ylabel('')
# Skip the E1 plot when
if not include_compartments:
ax.set_title(title)
continue
for j, (view, e1) in enumerate(e1_dict.items()):
ax1 = divider.append_axes("top", size="10%", pad=0.1, sharex = ax)
# Create stairs
x = np.zeros(2*chrom_start.size)
y = np.zeros(2*chrom_start.size)
x[0::2] = chrom_start/row.cooler_id.binsize
x[1::2] = chrom_end/row.cooler_id.binsize
y[0::2] = e1
y[1::2] = e1
ax1.fill_between(x, y, 0, where=(y > 0), color='tab:red', ec = 'None')
ax1.fill_between(x, y, 0, where=(y < 0), color='tab:blue', ec = 'None')
ax1.set_ylabel(view, rotation=0, ha='right', va='center')
ax1.set_ylim(-0.8, 0.8)
ax1.set_yticks([])
ax1.set_xticks([])
ax1.spines[:].set_visible(False)
ax1.set_title(title)
f.set_layout_engine('compressed')
plt.show()
Plotting matrices separately
Plotting function that makes separate plots in stead of mpl.subplots. This makes it compatible with YAML header subfigures and layout options for embedding into Quarto
def plot_for_quarto(
group: pd.DataFrame,
region: str | tuple[str, int, int] = 'chrX',
include_compartments: bool = True,
view_order: list[str] = ['10Mb', 'arms', 'full'],
figsize: tuple[float, float] = (6, 6),
col_vmin: float = 1e-3,
col_vmax: float = 0.1,
save_png_name: str = None
) -> None:
"""
Plot each cooler as a separate figure to allow Quarto YAML options for subfigure plots and captions.
"""
group = group.reset_index(drop=True)
# Scale figsize based on the number of coolers
figsize = (figsize[0]/group.shape[0], figsize[1])
# Set the region to a tuple if it is a string
if isinstance(region, str):
region = (region, 0, group.iloc[0].cooler_id.chromsizes[region])
# Define start and end for the matrix plot
start, end = region[1], region[2]
# Loop through each cooler (row) in the DataFrame
for row in group.itertuples():
# Create a new figure for each cooler
# Scale figsize
fig, ax = plt.subplots(figsize=figsize)
# Logtransform the colorscale
norm = LogNorm(vmin=col_vmin,vmax=col_vmax)
# Define the y-axis label
# title = f'{row.sname}{row.resolution}kb: {row.run}'
ylab = f'{row.resolution}kb'
# Extract some variables
e1_dict = {k: row.e1_dict[k] for k in view_order}
chrom_start = row.cooler_id.bins().fetch(region)['start'].values
chrom_end = row.cooler_id.bins().fetch(region)['end'].values
nbins = chrom_start.size # != row.nbins when subsetting chr
binsize = row.cooler_id.binsize # is used several times
# Plot the Hi-C matrix
im = ax.matshow(
row.cooler_id.matrix().fetch(region),
norm=norm,
cmap='fall',
extent=(0, nbins, nbins, 0)
)
ax.set_xlim(0, nbins)
ax.set_ylim(nbins, 0)
ax.set_ylabel(ylab)
ticks = np.linspace(0, nbins, 5)
ax.set_yticks(ticks)
ax.set_yticklabels([f'{(start/binsize+x)*binsize/1_000_000:.0f}' for x in ticks])
ax.xaxis.set_visible(False)
# Add a colorbar
divider = make_axes_locatable(ax)
cax = divider.append_axes("right", size="2%", pad=0.05)
plt.colorbar(im, cax=cax, label='corrected frequencies')
# Skip E1 plot if not including compartments
if include_compartments:
for view, e1 in e1_dict.items():
# subset the e1 to region (chrX:start-end)/binsize
start_bin = int(chrom_start[0]/binsize)
end_bin = int(chrom_start[-1]/binsize)
e1 = e1.iloc[start_bin:end_bin+1].reset_index(drop=True)
ax1 = divider.append_axes("top", size="10%", pad=0.05, sharex=ax)
# Create stairs for E1 values
x = np.zeros(2 * chrom_start.size)
y = np.zeros(2 * chrom_start.size)
x[0::2] = np.arange(chrom_start.size)
x[1::2] = np.arange(chrom_start.size)
y[0::2] = e1
y[1::2] = e1
ax1.fill_between(x, y, 0, where=(y > 0), color='tab:red', ec='None')
ax1.fill_between(x, y, 0, where=(y < 0), color='tab:blue', ec='None')
if len(e1_dict) > 1:
ax1.set_ylabel(view, rotation=0, ha='right', va='center', fontsize=7)
else:
ax1.set_ylabel('E1', rotation=0, ha='right', va='center', fontsize=7)
ax1.set_ylim(-0.8, 0.8)
ax1.set_yticks([])
ax1.set_xticks([])
ax1.spines[:].set_visible(False)
# # Set the figure title
# ax1.set_title(title)
fig.tight_layout()
if save_png_name:
plt.savefig(save_png_name, dpi=320)
# Display the figure for Quarto to capture
plt.show()
Plot intervals
Plot genomic intervals as tracks (up to 3)
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
from matplotlib.patches import Rectangle
from mpl_toolkits.axes_grid1 import make_axes_locatable
from genominterv import interval_intersect
def plot_regions(query=None, annot=None, intersect=None,
track_titles=['query', 'annot', 'intersect'],
figsize=(10, 1),
title='chrom',
alpha=0.8, lw=0):
chrom = annot['chrom'].unique()[0]
chromsizes = pd.read_csv('../data/rheMac10.filtered.chrom.sizes', sep='\t', header=None, names=['chrom', 'size'])
base_size = chromsizes[chromsizes['chrom'] == chrom]['size'].values[0]
annot_size = annot['end'].max()
query_size = query['end'].max()
chromsize = max(base_size, annot_size, query_size)
if title == 'chrom':
title = chrom
# Define the plot size
f, ax = plt.subplots(figsize=figsize, sharex=True)
ax.spines[:].set_visible(False)
# Plot the annot
# Iterate over each interval in the DataFrame
for start, end in zip(annot['start'], annot['end']):
rect = Rectangle((start, 0.1), width=end-start, height=0.9, color='tab:red', linewidth=lw, alpha=alpha)
ax.add_patch(rect)
ax.spines['bottom'].set_visible(True)
ax.set_ylabel(track_titles[1], rotation=0, labelpad=10, ha='right', va='center')
divider = make_axes_locatable(ax)
if query is not None:
qax = divider.append_axes("top", size="100%", pad=0.05, sharex=ax)
qax.xaxis.set_visible(False)
# Plot the query
for start, end in zip(query['start'], query['end']):
rect = Rectangle((start, 0.1), width=end-start, height=0.9, color='tab:blue', linewidth=lw, alpha=alpha)
qax.add_patch(rect)
qax.spines[:].set_visible(False)
qax.set_yticks([])
qax.set_title(title, loc='left')
qax.set_ylabel(track_titles[0], rotation=0, labelpad=10, ha='right', va='center')
if intersect is not None:
iax = divider.append_axes("bottom", size="100%", pad=0.05, sharex=ax)
# Invisible x-axis for 'annot' (intersect ie below)
ax.xaxis.set_visible(False)
# Plot the intersect
for start, end in zip(intersect['start'], intersect['end']):
rect = Rectangle((start, 0.1), width=end-start, height=0.9, color='tab:green', linewidth=lw, alpha=alpha)
iax.add_patch(rect)
iax.spines[:].set_visible(False)
iax.set_yticks([])
ax.spines['bottom'].set_visible(False)
iax.spines['bottom'].set_visible(True)
iax.set_ylabel(track_titles[2], rotation=0, labelpad=10, ha='right', va='center')
ax.set_yticks([])
ax.set_xlim(0, chromsize)
ticks = np.linspace(0, chromsize, num=5)
ax.set_xticks(ticks)
ax.set_xticklabels([f'{int(t/1e6)} Mbp' for t in ticks])
f.tight_layout()
plt.show()
#plt.tight_layout()
Make edges
def make_edges(df, resolution):
start_edge = pd.DataFrame({
'chrom': df['chrom'],
'start': df.apply(lambda x: max(x['start'] - resolution, 0), axis=1),
'end': df['start']+1*resolution,
'resolution': resolution,
})
end_edge = pd.DataFrame({
'chrom': df['chrom'],
'start': df['end']-1*resolution,
'end': df.apply(lambda x: min(x['end'] + resolution, chromsizes['chrX']), axis=1),
'resolution': resolution,
})
return interval_collapse(pd.concat([start_edge, end_edge]).sort_values(['chrom', 'start', 'end'])).assign(resolution=resolution)
Svedig tabel
all_tests = pd.read_csv('../results/all_tests.csv', index_col=0)
def svedig_tabel(orig_df, index, columns, values,
cmap='Reds', col_order=None, col_snames=None):
"""
Creates a styled pivot table from a DataFrame, filtering by significance,
applying a log transformation, and adding visual styling.
Parameters:
----------
orig_df : pd.DataFrame
The original DataFrame containing data to be processed.
index : str
Column name to use as the row index in the pivot table.
columns : str
Column name to use as the column headers in the pivot table.
values : str
Column name whose values populate the pivot table cells.
cmap : str, optional
Colormap to use for the background gradient styling (default is 'Reds').
Returns:
-------
pd.io.formats.style.Styler
A styled DataFrame object with the pivot table, including gradient-based
cell coloring, transparent NaN values, formatted numbers, and table styles.
"""
df = (orig_df
.assign(log10p=np.log10(all_tests.p))
.loc[(all_tests.p < 0.05)]
.pivot(index=index, columns=columns, values=values)
)
if col_order:
col_bool = [x in df.columns for x in col_order]
col_order = [x for x in col_order if x in df.columns]
df = df[col_order]
if col_snames:
col_snames = [col_snames[i] for i,x in enumerate(col_bool) if x]
df = df.rename(columns = {x:col_snames[i] for i,x in enumerate(df.columns.tolist())})
else:
df = df.rename(columns = {x:x.replace('_', '<br>') for x in df.columns.tolist()})
df = (df.style
.background_gradient(subset=df.columns, axis=None, cmap=cmap, vmin=0)
.map(lambda x: 'color: transparent; background-color: transparent' if np.isnan(x) else '')
.format('{:.3f}')
.set_table_styles(
{c: [{'selector': '',
'props': [('min-width', '20px')],
}] for c in df.columns}, overwrite=False
)
)
return dfprint("That should be all the setup :-)")That should be all the setup :-)Matrices / E1
E1: RS100/500
# Compare the compartments of the round_spermatid, 500kb, all views
res = ['100', '500']
name = ['round_spermatid']
run = ['recPE']
group = clr_df.query('name in @name and resolution in @res and run in @run')
plot_for_quarto(group)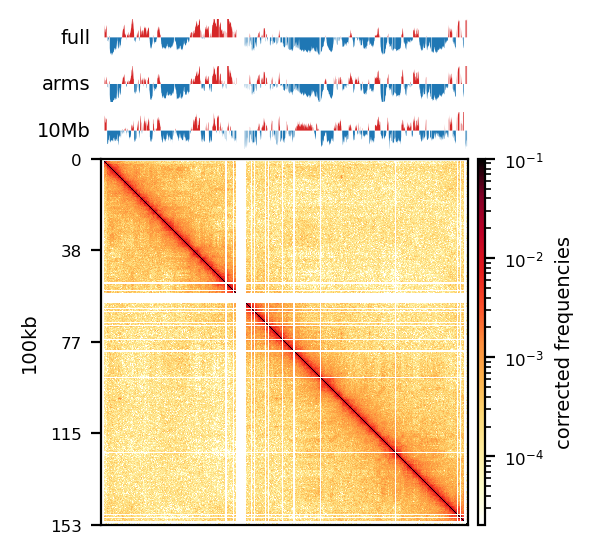
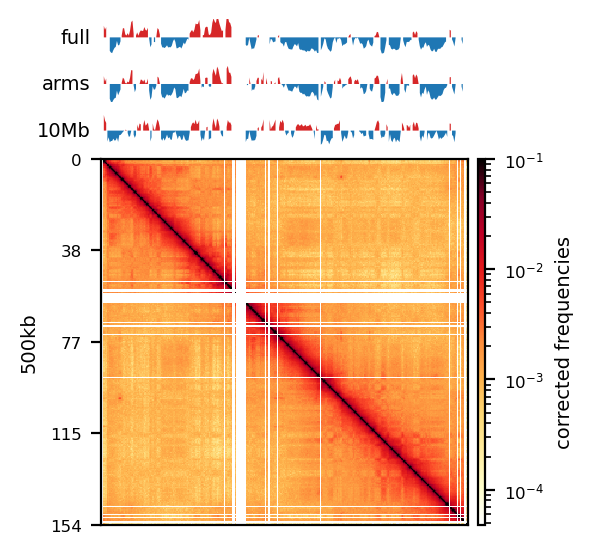
PE vs recPE (RS100kb)
# Compare PE and recPE compartments for round_spermatid, 100kb
group = clr_df.query('resolution == "100" and name == "round_spermatid"')
plot_grouped(group, figsize=(6, 6))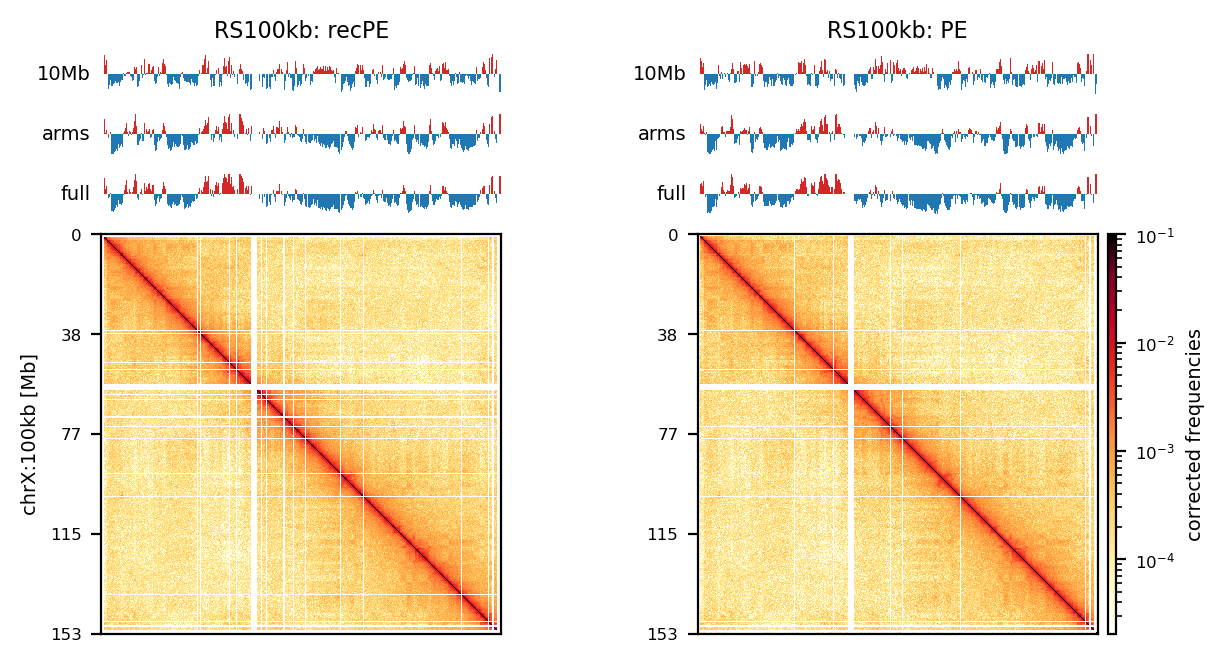
df = clr_df.query('resolution == "500" and name == "round_spermatid" and run == "PE"')
plot_grouped(df, include_compartments=False, figsize=(3, 3))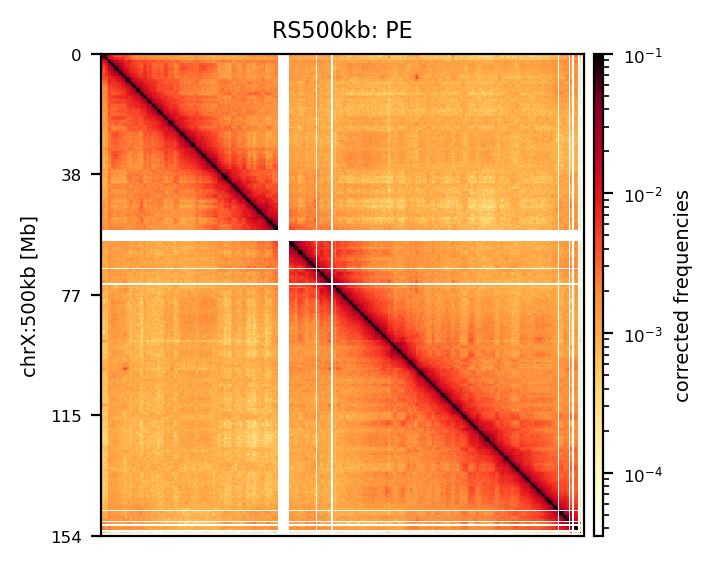
PE vs. recPE: RS100kb
df = clr_df.query('resolution == "100" and name == "round_spermatid"')
# Check the order of the group
#print(df[['name', 'resolution', 'run']].reset_index(drop=True))
plot_for_quarto(df, view_order=['10Mb','arms'], figsize=(5, 5))
start, end = 70_000_000, 78_000_000
region = ('chrX', start, end)
plot_for_quarto(df, region=region, include_compartments=True, view_order=['10Mb','arms'], figsize=(5, 5))
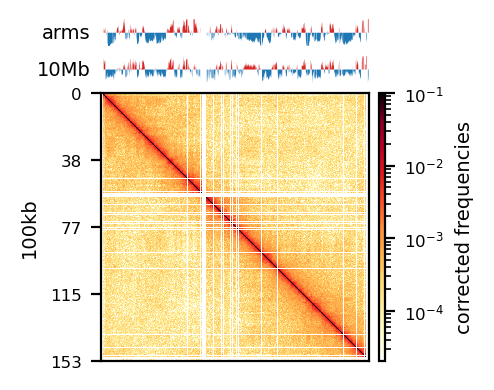
5unique: chrX:start-end
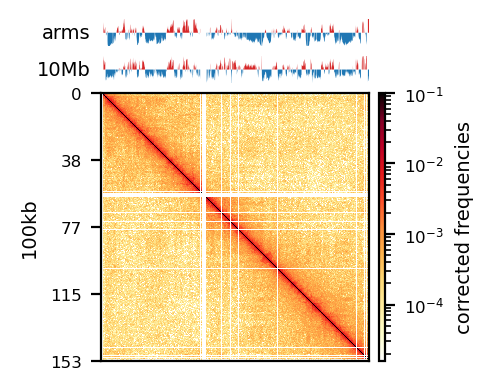
mask: chrX:start-end
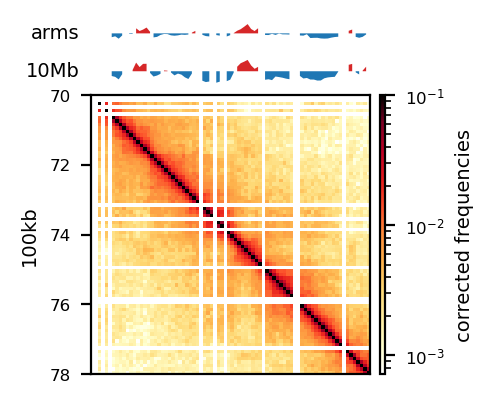
5unique: chrX:70Mb-78Mb
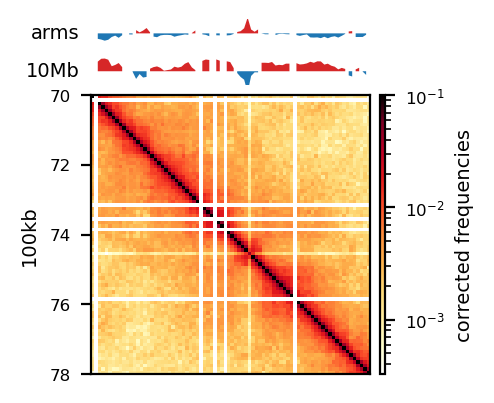
mask: chrX:70Mb-78Mb
Intervals
PE/recPE compare compartments (100kb)
from genominterv import interval_diff
### Plot the intervals and the diff between ###
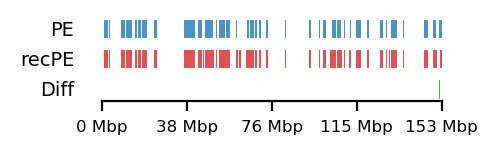
# Round Spermatid arms
query = comps['PE']['round_spermatid_100kb_arms']
annot = comps['recPE']['round_spermatid_100kb_arms']
plot_regions(query=query, annot=annot,
intersect=interval_diff(query, annot),
track_titles=['PE', 'recPE', 'Diff'],
figsize=(2.5, 0.8),
title=None)
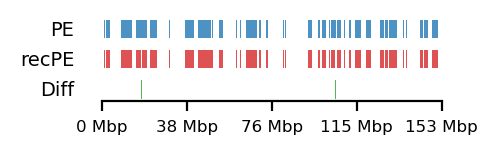
# Fibroblast arms
query = comps['PE']['fibroblast_100kb_arms']
annot = comps['recPE']['fibroblast_100kb_arms']
plot_regions(query=query, annot=annot,
intersect=interval_diff(query, annot),
track_titles=['PE', 'recPE', 'Diff'],
figsize=(2.5, 0.8),
title=None)
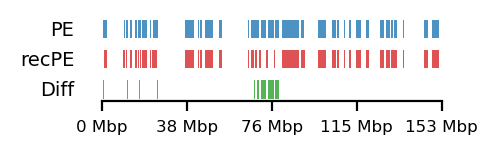
# Round Spermatid 10Mb
query = comps['PE']['round_spermatid_100kb_10Mb']
annot = comps['recPE']['round_spermatid_100kb_10Mb']
plot_regions(query=query, annot=annot,
intersect=interval_diff(query, annot),
track_titles=['PE', 'recPE', 'Diff'],
figsize=(2.5, 0.8),
title=None)
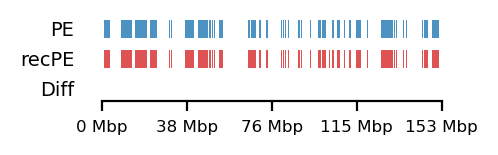
# Fibroblast 10Mb
query = comps['PE']['fibroblast_100kb_10Mb']
annot = comps['recPE']['fibroblast_100kb_10Mb']
plot_regions(query=query, annot=annot,
intersect=interval_diff(query, annot),
track_titles=['PE', 'recPE', 'Diff'],
figsize=(2.5, .8),
title=None)
from genominterv import interval_intersect
query = ech90
fb_ann_comp = comps['recPE']['fibroblast_100kb_arms']
rs_ann_comp = comps['recPE']['round_spermatid_100kb_arms']
fb_ann_edge = edges['recPE']['fibroblast_100kb_arms']
rs_ann_edge = edges['recPE']['round_spermatid_100kb_arms']
size=(3.2, 0.8)
# Fib and RS compartments
plot_regions(query=query, annot=fb_ann_comp,
intersect=interval_intersect(query, fb_ann_comp),
track_titles=['ECH90', 'A-Comp', 'Intersect'],
figsize=size,
title=None,
lw=0.3)
plot_regions(query=query, annot=rs_ann_comp,
intersect=interval_intersect(query, rs_ann_comp),
track_titles=['ECH90', 'A-Comp', 'Intersect'],
figsize=size,
title=None,
lw=0.3)
# Fib and RS edges
plot_regions(query=query, annot=fb_ann_edge,
intersect=interval_intersect(query, fb_ann_edge),
track_titles=['ECH90', 'Edges', 'Intersect'],
figsize=size,
title=None,
lw=0.3, alpha=1)
plot_regions(query=query, annot=rs_ann_edge,
intersect=interval_intersect(query, rs_ann_edge),
track_titles=['ECH90', 'Edges', 'Intersect'],
figsize=size,
title=None,
lw=0.3, alpha=1)
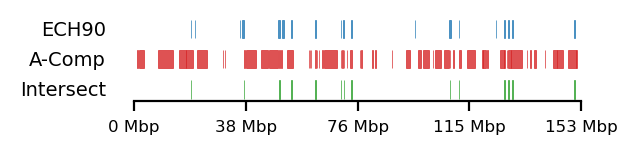
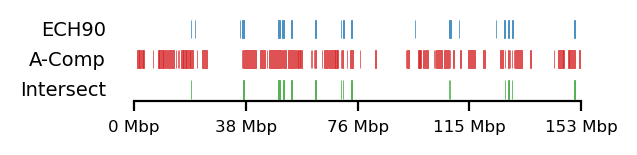
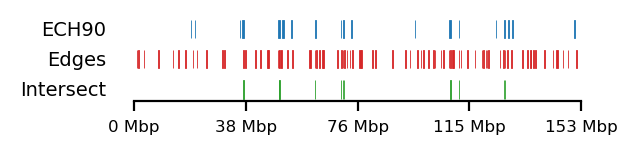
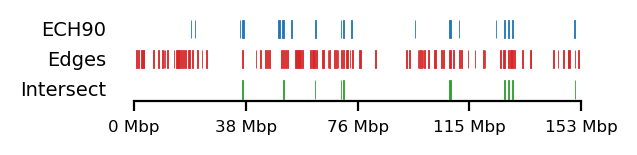
Visualize ECH enrichment
from genominterv import interval_diff, interval_intersect, interval_union
complist = [comps['recPE']['round_spermatid_100kb_arms'],
comps['recPE']['fibroblast_100kb_arms']]
edgelist = [edges['recPE']['round_spermatid_100kb_arms'],
edges['recPE']['fibroblast_100kb_arms']]
for comp, edge in zip(complist, edgelist):
int_comp = interval_intersect(ech90, comp)
int_edge = interval_intersect(ech90, edge)
diff_int_compedge = interval_diff(int_edge, int_comp)
plot_regions(int_comp, int_edge, diff_int_compedge,
track_titles=['CompInt', 'EdgeInt', 'DiffInt'],
title="ECH90 intersect",
figsize=(3.2, 1),
alpha=1, lw=0.5)
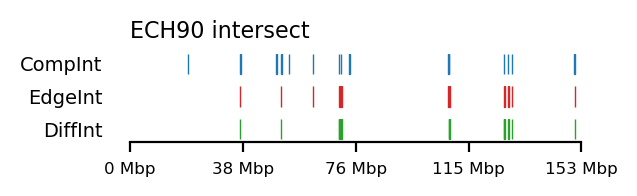
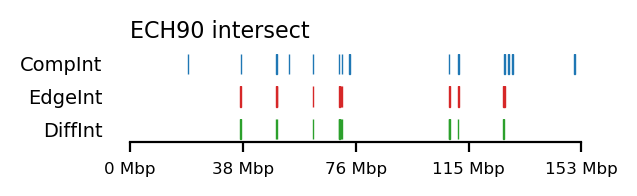
All 3 sets
plot_regions(ech90, high_olive, high_hama,
track_titles=['ECH90', r"$\it{P.anubis}$", r"$\it{P.hama}$"],
figsize=(5, 0.8),
title=None,
lw=0.5, alpha=0.8)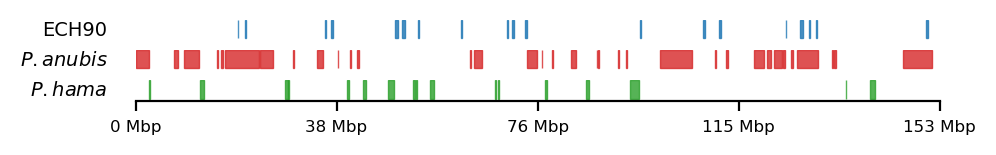
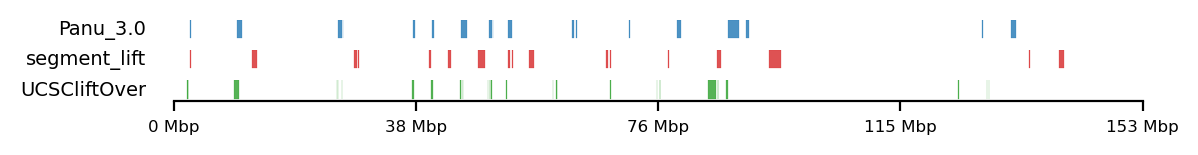
LiftOver comparison
track0 = [olive_panu3, hama_panu3]
track1 = [high_olive, high_hama]
track2 = [high_olive_ucsc, high_hama_ucsc]
for t0,t1,t2 in zip(track0, track1, track2):
plot_regions(t0,t1,t2,
track_titles=['Panu_3.0', 'segment_lift','UCSCliftOver'],
# title='High Olive Ancestry Intervals',
title=None,
figsize=(6, 0.8),
alpha=0.8, lw=0.1)
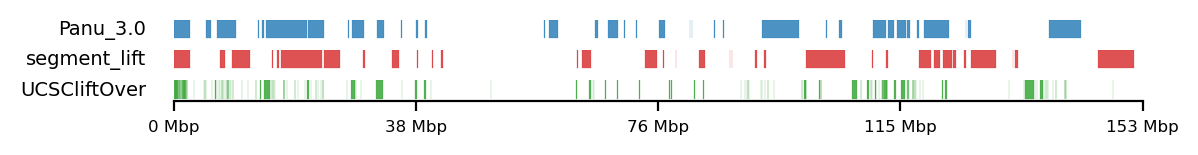
Baboon-rs100-intersect
# Plot
size = (3.2, 0.8)
edge_rs = edges['recPE']['round_spermatid_100kb_arms']
for name,group in baboon_dict.items():
plot_regions(query=edge_rs, annot=group,
intersect=interval_intersect(edge_rs, group),
#intersect=ech90,
track_titles=['RS100', fr"$\it{{{name}}}$", 'Intersect'],
figsize=size,
title=None,
lw=0.3, alpha=0.9)
edge_rs = edges['recPE']['round_spermatid_100kb_10Mb']
for name,group in baboon_dict.items():
plot_regions(query=edge_rs, annot=group,
intersect=interval_intersect(edge_rs, group),
#intersect=ech90,
track_titles=['RS100', fr"$\it{{{name}}}$", 'Intersect'],
figsize=size,
title=None,
lw=0.2, alpha=1)
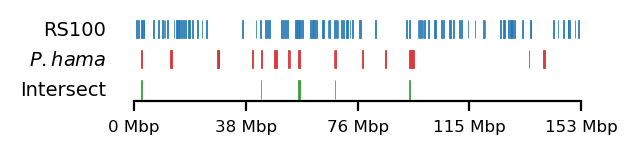
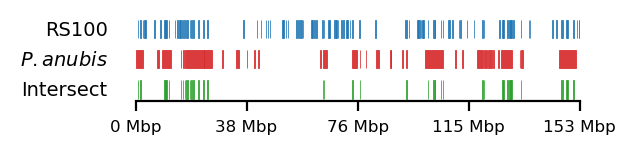
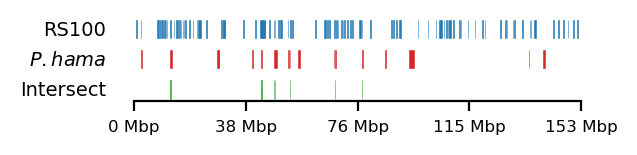
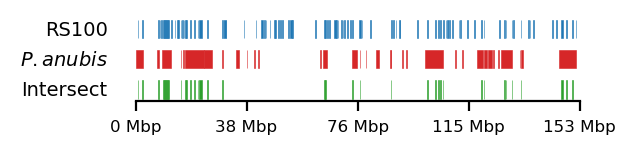
Table: Test 1bp limits
all_tests.head() #.query('query == "hama_edge_1bp"')| tissue | pc_scale | query | annot | test | value | p | -log10p | |
|---|---|---|---|---|---|---|---|---|
| 0 | fibroblast | arms | ECH90 | comp_edge_1bp | jaccard | 0.000002 | 0.01656 | 1.780940 |
| 1 | fibroblast | arms | hama_edge_1bp | comp_edge_1bp | proximity | 0.221717 | 0.88421 | 0.053445 |
| 2 | fibroblast | arms | olive_edge_1bp | comp_edge_1bp | proximity | 0.243643 | 0.67235 | 0.172405 |
| 3 | fibroblast | arms | olivehama_edge_1bp | comp_edge_1bp | proximity | 0.243643 | 0.74099 | 0.130188 |
| 4 | fibroblast | 10Mb | ECH90 | comp_edge_1bp | jaccard | 0.080105 | 0.02146 | 1.668370 |
tissue_order = ['fibroblast', 'spermatogonia', 'pachytene_spermatocyte', 'round_spermatid', 'sperm']
tissue_sname = ['Fb', 'Spa', 'Pac', 'RS', 'Sperm']
t0 = (svedig_tabel(all_tests
.map(lambda x: x.replace('_edge_1bp', '') if isinstance(x, str) else x)
.rename(columns={'pc_scale': 'viewframe'}),
index=['viewframe', 'test','query'],
columns=["tissue"],
col_order = tissue_order,
col_snames = tissue_sname,
values='p',
cmap='Reds_r')
)
t1 = (svedig_tabel(
all_tests
.map(lambda x: x.replace('_edge_1bp', '') if isinstance(x, str) else x)
.query('pc_scale == "arms"'),
index=['test','query'],
columns=["tissue"],
col_order = tissue_order,
col_snames = tissue_sname,
values='p',
cmap='Reds_r')
#.relabel_index(labels=[lbl for lbl in tissue_sname if lbl in t1.columns], axis=1)
)
t2 = (svedig_tabel(all_tests
.map(lambda x: x.replace('_edge_1bp', '') if isinstance(x, str) else x)
.query('pc_scale == "10Mb"'),
index=['test','query'],
columns=["tissue"],
col_order = tissue_order,
col_snames = tissue_sname,
values='p',
cmap='Reds_r')
)
display(t0)| tissue | Fb | Spa | Pac | RS | Sperm | ||
|---|---|---|---|---|---|---|---|
| viewframe | test | query | |||||
| 10Mb | jaccard | ECH90 | 0.021 | 0.006 | nan | nan | 0.048 |
| proximity | olive | nan | nan | nan | nan | 0.016 | |
| olivehama | nan | 0.019 | 0.012 | nan | 0.001 | ||
| arms | jaccard | ECH90 | 0.017 | 0.006 | 0.005 | 0.028 | nan |
| proximity | olivehama | nan | nan | 0.036 | nan | nan |
tissue_order = ['fibroblast', 'spermatogonia', 'pachytene_spermatocyte', 'round_spermatid', 'sperm']
tissue_sname = ['Fb', 'Spa', 'Pac', 'RS', 'Sperm']
df = (all_tests
.map(lambda x: x.replace('_edge_1bp', '_1bp') if isinstance(x, str) else x)
.assign(log10p=np.log10(all_tests.p))
.loc[(all_tests.p < 0.05)]
.pivot(index=['pc_scale', 'query', 'test' ], columns=["tissue"], values="p").
filter(tissue_order)
)
df = df.rename(columns = {x:tissue_sname[i] for i,x in enumerate(df.columns.tolist())})
df.columns.name = None
df = df.reset_index().fillna('-')
df = df.rename(columns={'pc_scale': 'View', 'query': 'Query', 'test': 'Test'})
df.style.hide(axis='index')| View | Query | Test | Fb | Spa | Pac | RS | Sperm |
|---|---|---|---|---|---|---|---|
| 10Mb | ECH90 | jaccard | 0.021460 | 0.006110 | - | - | 0.047500 |
| 10Mb | olive_1bp | proximity | - | - | - | - | 0.016140 |
| 10Mb | olivehama_1bp | proximity | - | 0.018790 | 0.012300 | - | 0.001280 |
| arms | ECH90 | jaccard | 0.016560 | 0.006120 | 0.005340 | 0.027690 | - |
| arms | olivehama_1bp | proximity | - | - | 0.035640 | - | - |
Figures for the defence
%config InlineBackend.figure_formats = ['retina']group = clr_df.query('name == "sperm" and resolution == "500" and run == "recPE"')
slide_path = "../slides/images/hic-example.png"
plot_for_quarto(group, view_order=['10Mb'],
col_vmin=1e-4, col_vmax=0.5,
save_png_name=slide_path)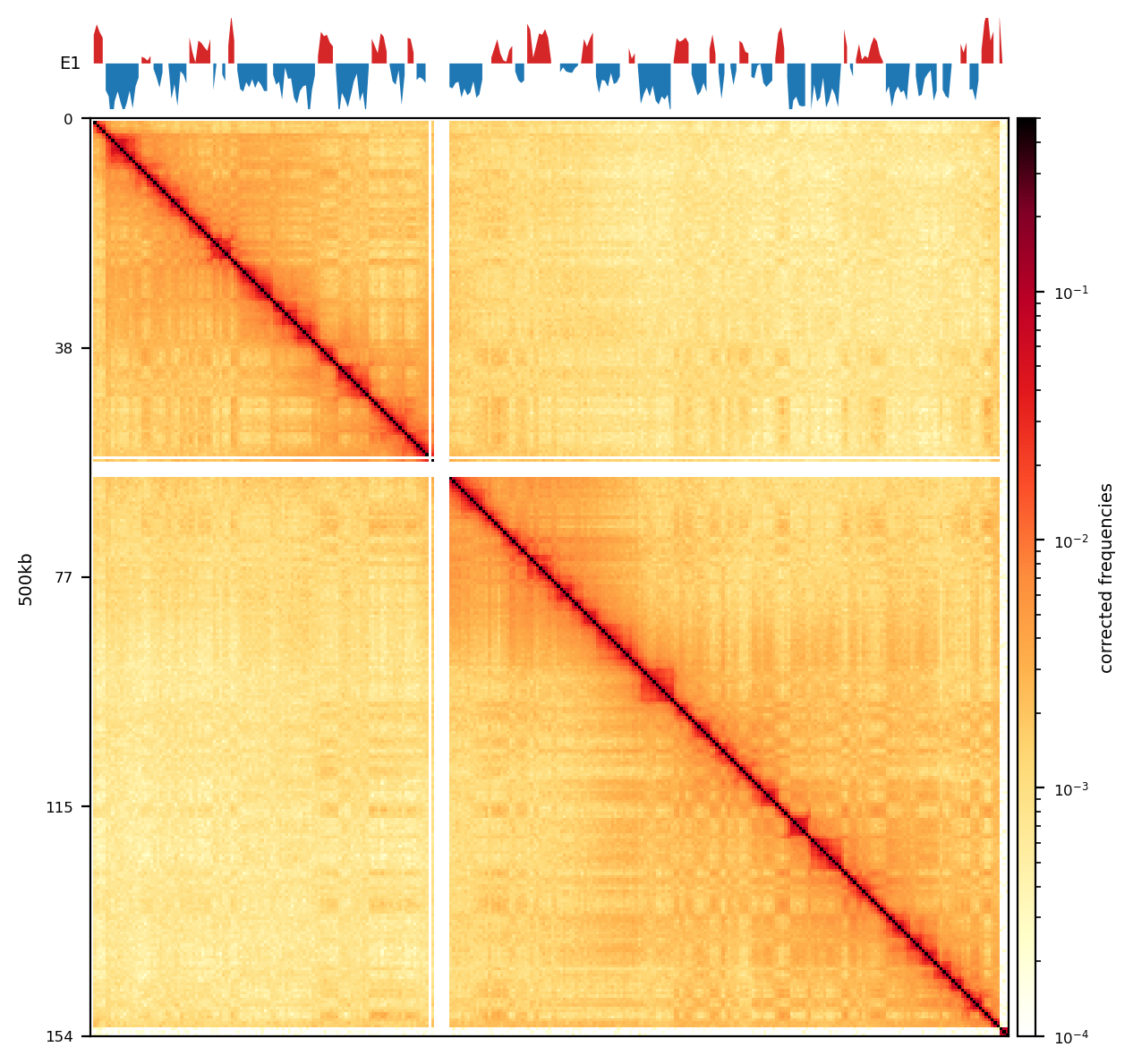
Introduce selected regions
plot_regions(ech90, high_olive, high_hama,
track_titles=['ECH90', r"$\it{P.anubis}$", r"$\it{P.hama}$"],
figsize=(10, 1.5),
title=None,
lw=0.5, alpha=0.8)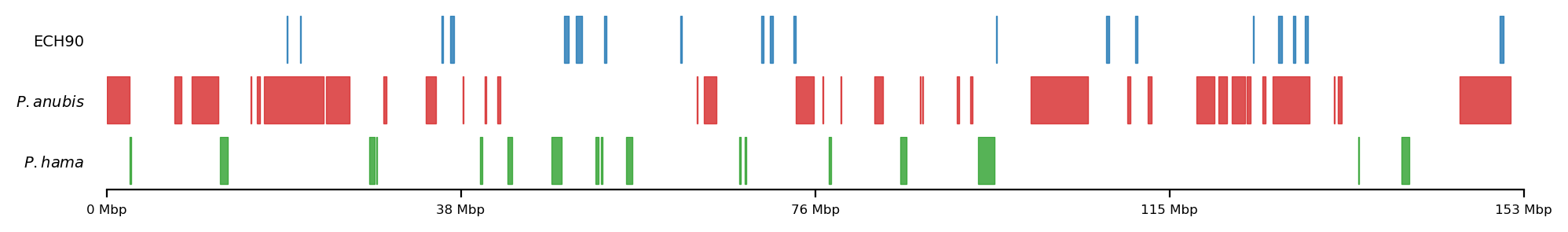
Compare liftover
from genominterv import interval_union
track0 = [olive_panu3, hama_panu3]
track1 = [high_olive, high_hama]
track2 = [high_olive_ucsc, high_hama_ucsc]
track0 = [interval_union(olive_panu3, hama_panu3)]
track1 = [interval_union(high_olive, high_hama)]
track2 = [interval_union(high_olive_ucsc, high_hama_ucsc)]
for t0,t1,t2 in zip(track0, track1, track2):
plot_regions(t0,t1,t2,
track_titles=['Panu_3.0', 'segment_lift','UCSCliftOver'],
# title='Baboon Intervals',
title=None,
figsize=(10, 1.5),
alpha=0.8, lw=0.2)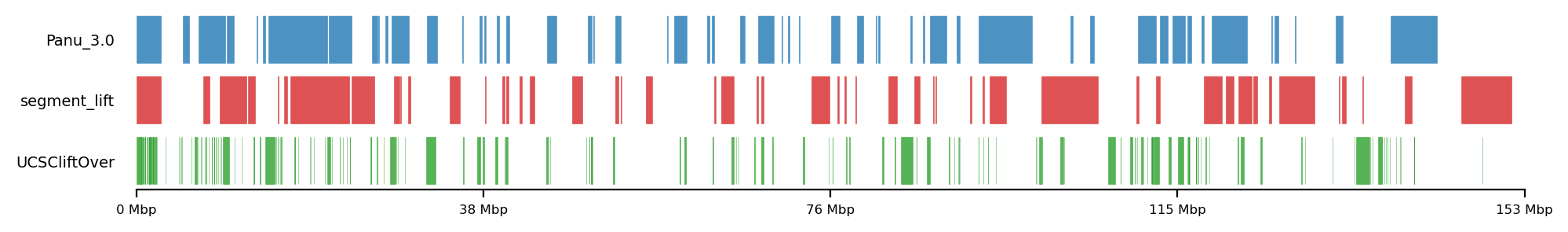
ECH S100 limit intersect
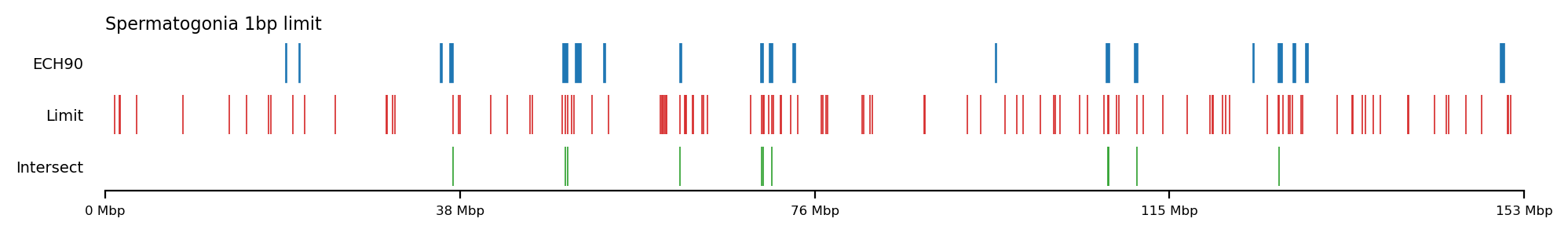
annot = make_edges(comps['recPE']['fibroblast_100kb_arms'], 2)
query = ech90
plot_regions(query=query, annot=annot,
intersect=interval_intersect(query, annot),
track_titles=['ECH90', 'Limit', 'Intersect'],
figsize=(10, 1.5),
title="Spermatogonia 1bp limit",
lw=0.6, alpha=1)
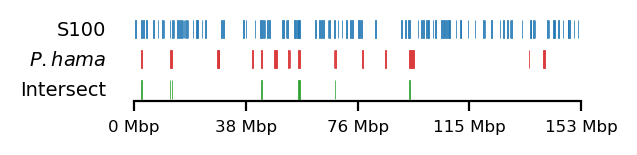
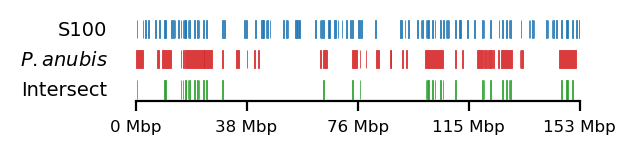
# Plot
size = (3.2, 0.8)
edge_rs = edges['recPE']['sperm_100kb_arms']
for name,group in baboon_dict.items():
plot_regions(query=edge_rs, annot=group,
intersect=interval_intersect(edge_rs, group),
#intersect=ech90,
track_titles=['S100', fr"$\it{{{name}}}$", 'Intersect'],
figsize=size,
title=None,
lw=0.3, alpha=0.9)
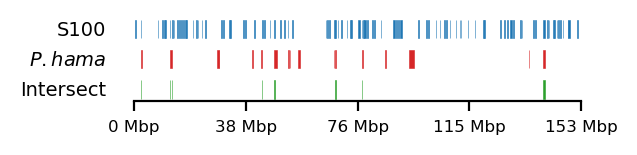
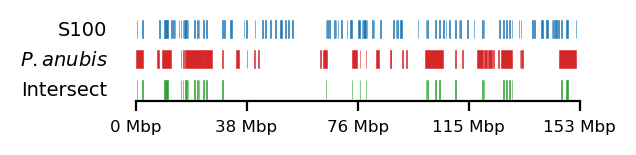
edge_rs = edges['recPE']['sperm_100kb_10Mb']
for name,group in baboon_dict.items():
plot_regions(query=edge_rs, annot=group,
intersect=interval_intersect(edge_rs, group),
#intersect=ech90,
track_titles=['S100', fr"$\it{{{name}}}$", 'Intersect'],
figsize=size,
title=None,
lw=0.2, alpha=1)