33 Read mapping
Next-generation sequencing (NGS) has become extremely popular among geneticists and bioinformaticians in recent decades. Three of the biggest sequencing companies are Illumina, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT). The three companies use different technologies to sequence DNA, and they each have pros and cons. Illumina reads are relatively short (150 bp) but are also the cheapest solution. PacBio produces long, accurate reads (~15 kb) but is more than twice as expensive as Illumina. ONT can produce extremely long reads (60 kb) but suffers from low-quality base calls.
The choice of sequencing technique is highly dependent on the specific research question, so it is worth understanding the capabilities of the methods before choosing how the DNA should be sequenced.
33.1 HIFI reads
In this exercise, you will work with PacBio HiFi reads. Currently, this sequencing technique is the most accurate of the NGS technologies. The sequencing method is shown in the below figure. The biggest strength of this sequencing technique is that the reads are read multiple times. Every time the read is read, a subread is produced, and the final HiFi read is the consensus of all the subreads. This approach eliminates most of the sequencing errors found in the subreads.
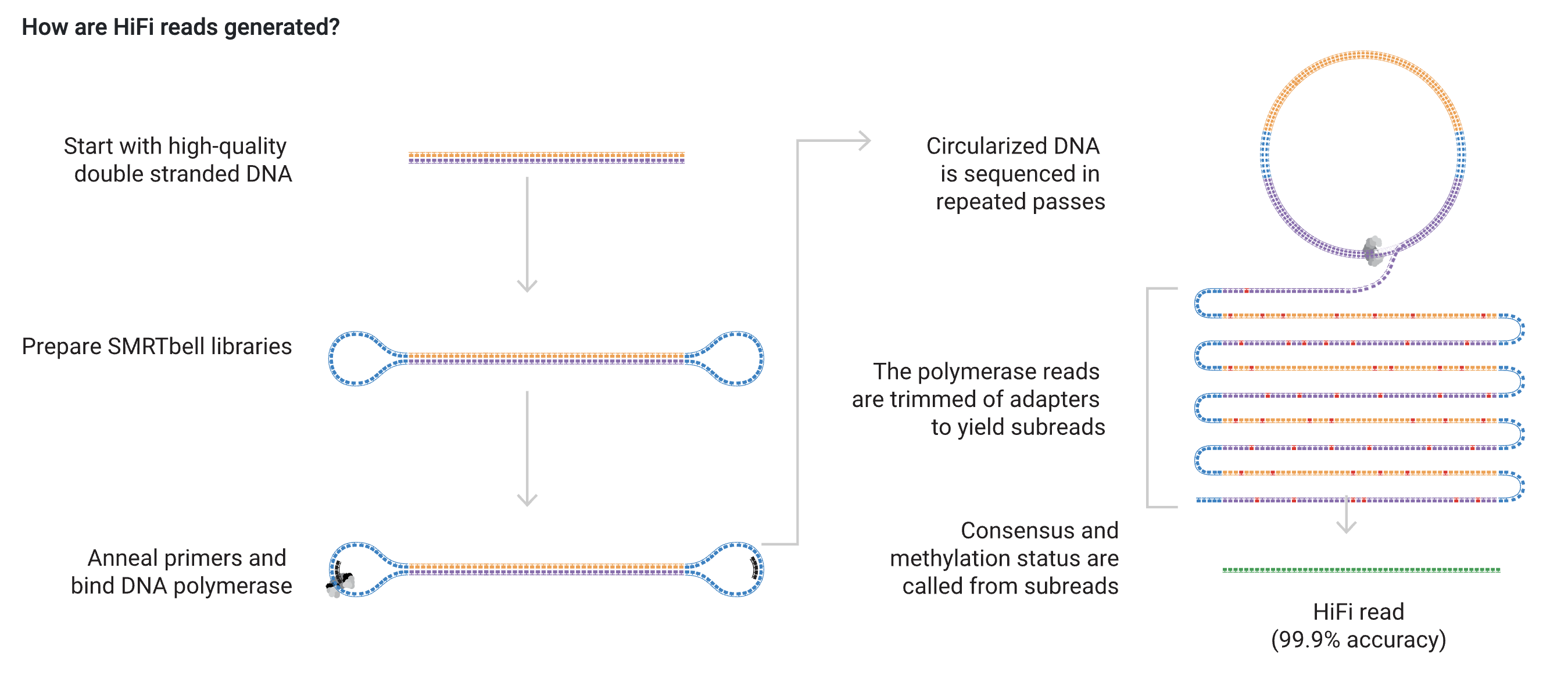
In this exercise, you will work on a 3.5 Mb contig from an unknown chromosome and tissue. The reads are PacBio HiFi reads, and in this sample, the average read length is approximately 10 kb. We can use the IGV (Integrative Genomics Viewer) program to visualize bam files containing the reads.
Exercise 33.1.0.1
Go to the IGV download page and download the program that fits your computer. You should also download the four files found on Brightspace. Once downloaded, you are ready to load the read data in IGV. In the top panel, click Genomes > Load Genome from file and then load the file called contig_1.fa. Now you should be able to see that the contig is 3534 kb. Next, click on File > Load from file in the top panel and load the file called contig_1.bam (if you get something like “index error”, ensure that all your files are named correctly, no spaces in the file names and that they are all placed in the same folder). You can zoom in and out in the top right corner of your screen. Zoom in until you see a window size of 23 kb. If you want to go to a specific region, you can change the coordinates in the white box next to the contig name. The format is like the UCSC Genome Browse (chr:start-end). Drag the data track to see different areas on the contig. If you encounter problems, watch this five-minute video. Play around for five minutes to understand what you are looking at.
- What are the purple ’I’s and the black dots,
- How does the coverage vary?
- Try to zoom in as much as you can. What information appears in the lower panel?
- Click on one of the reads; what kind of information do you see? Some of the lines are obvious, but some of them are more tricky. Find some of the tags on this page. You should focus on MM, np, rq, clipping, and QV tags:
MM: This tag denotes the probability of a cytosine being methylated when a guanine follows the cytosine. The dinucleotide CG is often referred to as a CpG site. These sites are unique due to their elevated mutation rate, but that topic will be another time.
NP: The np tag tells you how many times a read is read in the PacBio machine. If a read is read once or twice, it often results in low base qualities. In contrast, if the read is read 15-20 times, we are more confident that the base calls are correct.
QV: Denotes the quality of the base calling. It ranges from 1-93 on the Phred scale (https://genome.sph.umich.edu/wiki/Phred_scale). The tag is highly correlated with the np tag because the more a read is read, the higher the base calling quality.
RQ: This tag shows the overall quality of the entire read.
Clipping: If a proportion of the read does not fit the reference sequence, that proportion can be masked/clipped. Because of this masking, the read will look shorter in IGV.
Exercise 33.1.0.2
Go to: ptg000001l:1003200-1007274. What is the maximum coverage in this region? There is a read with a red arrowhead at the end of the reads. What does that mean (Hint: Look at the tags and compare them to the rest of the reads; one of the tags is different)? How does your conclusion relate to the length of the read that is shown? Right-click on your screen, and you will see a lot of options. Click on Quick consensus mode. Can you see a difference in some of the reads? You will see two red and green lines on one of the reads. To find out what the lines represent, try to zoom in on these positions and click on each line.
- What does the colored line represent?
- Which base is highlighted in green, and which is highlighted in red?
- As we know, there are four nucleotides. Find out which colors are related to the last two nucleotides (You should zoom out to find these).
- Now go back to
ptg000001l:1003200-1007274. You have probably figured out that the colored lines represent a mismatch in the alignment. These mismatches could be rare but true mutations, but they could also be sequencing errors. Use your knowledge about the np tag and QV tag to determine whether the mismatches are due to sequencing errors or true mutations. (Hint: Compare the tags with the reads that do not have mismatches)
Exercise 33.1.0.3
Now that you are an expert in IGV, we are ready for more advanced stuff. Pack your suitcase and travel to ptg000001l:2879315-2879495. Right-click and click on Hide small indels.
- How many mismatches, insertions, and deletions do you see? You should also include the length of the indels.
- You might have noticed that some of the reads have turned white. What does the white color mean (Hint: Look at the tags)?
Exercise 33.1.0.4
In one of the previous exercises, you found positions where only one read contained the mismatch. This mismatch is often referred to as a singleton. Base quality (QV) is essential when determining whether the mismatch is a mutation or a sequencing error. In this exercise, you look at another measure called mapping quality (MAPQ). This quality measure ranges from 0 to 60, telling you how unique a read is mapped. If the mapping quality is 0, the read fits equally well in another part of the genome. In this exercise, you will also see what highly frequent single nucleotide polymorphisms (SNPs) look like. Lastly, you will learn to think critically about the coverage and understand how high coverage relates to poor mapping quality.
Go to ptg000001l:3178259-3178531.
- What is the maximum coverage in this region, and how does that compare to the values from the previous exercises?
You should be able to see four SNPs. Click on the colored bars in the coverage panel.
- What is the count for each nucleotide at the SNP position?
- What do the numbers in the parenthesis tell you?
- Try to click on five different reads. What is the MAPQ for each of these reads (write them down)?
- Compare the five values with the MAPQ of five reads found in the previous region (
ptg000001l:2879315:2879495). Are they different? Which region do you trust more?
Exercise 33.1.0.5
Place your cursor on “Color alignments by,” and choose “base modification (5mc)”. What do you see?
Zoom in on one of the blue/red boxes and click some. What do they show you? (Hint: Look at the sequence. Which two bases do you see for all the blue/red colors?)
Go to ptg000001l:910,755-912,644. You might have noticed that the blue/red color represents the probability of the C being methylated. In this region, you can see that the blue color dominates and that the concentration of CpG sites is high. To see this more clearly, zoom out by clicking the minus button five times. The region looks unique, so let’s investigate it a bit more.
Exercise 33.1.0.6
Next to where you put your coordinates, there is a blue square with a red line on it. Click this symbol and move your cursor to the left side of your alignment. Your cursor should look like a “plus” sign. Click on the alignment, then move the cursor to the right side of the screen and click again. The region should now be highlighted by a red line, like in the figure. Click the red line, and click “Copy sequence”. Go to the Bast home page, paste the sequence, and press ‘BLAST’.
- Which chromosome is the sequence from?
- The sequence aligns 100 % with a gene called KDM5D. Try to explain why it is likely that the blue cluster in IGV represents a gene.
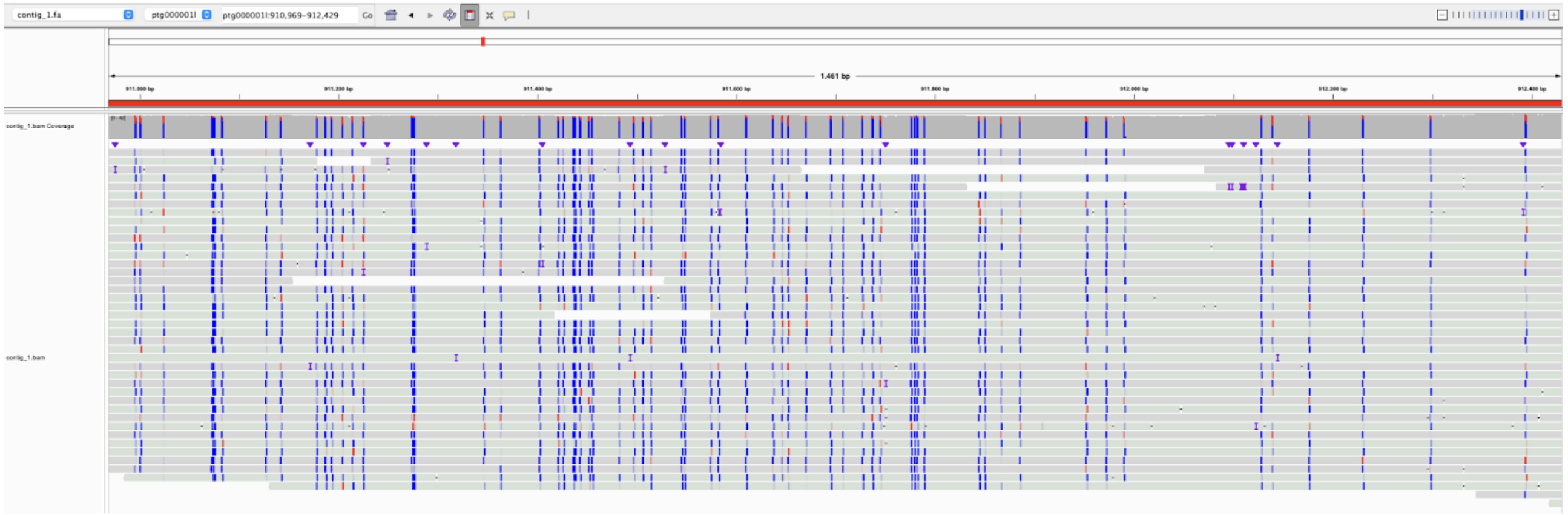
Exercise 33.1.0.7
Go to `ptg000001l:1,855,223-1,901,969``. You should see a region with a high number of CpGs, but the base modification differs between the reads. Discuss what causes this pattern.
Exercise 33.1.0.8
It is time for the grand finale. Go to the end destination, which is found at ptg000001l:243,727-244,325.
- Do you think the region contains a gene? Explain why/why not
- Highlight the region, and BLAST the sequence.
- Which tissue do you think the DNA comes from?
- If you have time, discover more interesting patterns in the data set.
Project files
Download the files you need for this project: